- Search Menu
- Editor's Choice
- Advance articles
- Review Series
- Virtual Issues
- Author Guidelines
- Submission Site
- Open Access Options
- Self-Archiving Policy
- Call for Papers
- About Clinical & Experimental Immunology
- About the British Society for Immunology
- Editorial Board
- Early Career Researcher Editorial Board
- Advertising & Corporate Services
- Publishing with us
- Journals on Oxford Academic
- Books on Oxford Academic


Article Contents
Other articles published in this series, introduction.
- < Previous
Myasthenia gravis: an update for the clinician
- Article contents
- Figures & tables
- Supplementary Data
J P Sieb, Myasthenia gravis: an update for the clinician, Clinical and Experimental Immunology , Volume 175, Issue 3, March 2014, Pages 408–418, https://doi.org/10.1111/cei.12217
- Permissions Icon Permissions
This paper provides a thorough overview of the current advances in diagnosis and therapy of myasthenia gravis (MG). Nowadays the term ‘myasthenia gravis’ includes heterogeneous autoimmune diseases, with a postsynaptic defect of neuromuscular transmission as the common feature. Myasthenia gravis should be classified according to the antibody specificity [acetylcholine, muscle-specific receptor tyrosine kinase (MuSK), low-density lipoprotein receptor-related protein 4 (LRP4), seronegative], thymus histology (thymitis, thymoma, atrophy), age at onset (in children; aged less than or more than 50 years) and type of course (ocular or generalized). With optimal treatment, the prognosis is good in terms of daily functions, quality of life and survival. Symptomatic treatment with acetylcholine esterase inhibition is usually combined with immunosuppression. Azathioprine still remains the first choice for long-term immunosuppressive therapy. Alternative immunosuppressive options to azathioprine include cyclosporin, cyclophosphamide, methotrexate, mycophenolate mofetil and tacrolimus. Rituximab is a promising new drug for severe generalized MG. Emerging therapy options include belimumab, eculizumab and the granulocyte– macrophage colony-stimulating factor. One pilot study on etanercept has given disappointing results. For decades, thymectomy has been performed in younger adults to improve non-paraneoplastic MG. However, controlled prospective studies on the suspected benefit of this surgical procedure are still lacking. In acute exacerbations, including myasthenic crisis, intravenous immunoglobulin, plasmapheresis and immunoadsorption are similarly effective.
Paraneoplastic neurological syndromes. Clinical and Experimental Immunology 2014, 175: 336–48.
Diagnosis, pathogenesis and treatment of myositis: recent advances. Clinical and Experimental Immunology 2014, 175: 349–58.
Disease-modifying therapy in multiple sclerosis and chronic inflammatory demyelinating polyradiculoneuropathy: common and divergent current and future strategies. Clinical and Experimental Immunology 2014, 175: 359–72.
CLIPPERS: chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. Review of an increasingly recognized entity within the spectrum of inflammatory central nervous system disorders. Clinical and Experimental Immunology 2014, 175: 385–96.
Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clinical and Experimental Immunology 2014, 175: 397–407.
Monoclonal antibodies in treatment of multiple sclerosis. Clinical and Experimental Immunology 2014, 175: 373–84.
Cerebral vasculitis in adults: what are the steps in order to establish the diagnosis? Red flags and pitfalls. Clinical and Experimental Immunology 2014, 175: 419–24.
Multiple sclerosis treatment and infectious issues: update 2013. Clinical and Experimental Immunology 2014, 175: 425–38.
Disorders of neuromuscular transmission can be of immunological, toxic or genetic origin. Among these rare disorders, myasthenia gravis is the most frequent. The clinical hallmark of myasthenia (MG) gravis is a fluctuating pronounced weakness limited to the voluntary muscles. Characteristically, muscular exertion increases the myasthenic weakness. It is a generalized disorder that often manifests initially as focal weakness. Eye muscle weakness at the onset of MG is evident in the vast majority of patients resulting in diplopia and ptosis. If the weakness is limited to the ocular muscles, it is designated ‘ocular myasthenia’. Oropharyngeal weakness may cause difficulties in articulation, chewing and swallowing. In generalized myasthenia gravis, limb girdle weakness is typically more pronounced in the proximal than in the distal muscle groups. Myasthenic crisis is the life-threating exacerbation of MG due to weakness of respiratory muscles and swallowing difficulties.
Surprisingly, epidemiological studies from Canada, Italy and Japan have observed an increasing frequency of MG in the elderly during recent decades [ 1–3 ]. In British Columbia, the annual number of first-time anti-acetylcholine receptor (AChR)-positive MG cases increased from 21·4/year/million during 1984–88 to 52·9 during 2004–08 in the elderly with an age of at least 65 years [ 1 ]. This international phenomenon might be a result of an increased awareness among medical doctors considering more frequently the diagnostic possibility of MG in the elderly.
Nowadays the term ‘myasthenia gravis’ describes a heterogeneous group of autoimmune diseases with a postsynaptic defect of neuromuscular transmission. These myasthenic syndromes can be divided according to the following categories with distinct clinical features and specific therapeutic needs:
course type:
ocular (in approximately 20% of MG patients)
oropharyngeal or generalized
age of onset:
start before puberty
early onset before the age of 50 years
late onset after the age of 50 years [ 4 ]
antibody specificity:
anti-muscle-specific receptor tyrosine kinase (MuSK)
anti-low-density lipoprotein receptor-related protein 4 (LRP4)
seronegative MG
pathology of the thymus
normal/atrophic thymus pathology
paraneoplastic occurrence associated with thymoma
In about 50% of those patients with ocular MG and in at least 10–15% with a generalized disease, the testing for autoantibodies to the AChR gives negative results. Some of these ‘seronegative’ patients have low-affinity antibodies to AChR that cannot be detected in standard solution phase assays, but can be detected in a novel method developed by a British group [ 5 ]. To increase sensitivity, recombinant AChR subunits were expressed with the clustering protein, rapsyn, in human embryonic kidney cells. Antibody binding to the AChR clustered at cell surface is visualized by immunofluorescence. Using this method, the British authors detected AChR antibodies to rapsyn-clustered AChR in two-thirds of sera previously negative for binding to AChR in solution. Unfortunately, this laboratory method for detection of low-affinity anti-AChR antibodies has not been introduced commercially, and therefore it is not generally available for diagnostic purposes.
However, the seronegativity present in some MG patients does not result from the insufficient sensitivity of the applied laboratory method. In this subset of seronegative patients, myasthenic weakness comes from autoimmune processes directed to postsynaptic targets distinct from the AChR. Antibodies against MuSK are present in approximately 40% of seronegative cases with generalized MG, and in another portion of myasthenic patients, antibodies against LRP4 are detectable. MuSK and LRP4 are not involved directly in the neuromuscular transmission, but in the end-plate maturation. The membrane protein LRP4 is the receptor of glycoprotein agrin, which is released by the nerve terminal. Binding of agrin to LRP4 stimulates myotubes to form clusters of AChRs linked via rapsyn [ 6 ]. The MuSK is involved in this cellular signal processing. The anti-MuSK antibody-positive MG was identified in 2001 [ 7 ]. Little is still known about the MG with anti-LRP4 anti-antibodies. This subtype was described independently in 2011 by two groups from Japan and Germany, respectively, [ 8 , 9 ]. The anti-LRP4 MG occurs predominately in middle-aged women. Overall, the clinical phenotype resembles closely that of anti-AChR-positive MG. It should be emphasized that even after the identification of anti-LRP 4 some MG patients are ‘triple seronegative’.
Anti-MuSK antibody-positive MG
There are several clinically important differences between this type of MG and the anti-AChR antibody-positive type: only rarely is muscular weakness limited to the eye muscles, which are affected predominately in the majority of patients with anti-AChR antibody-positive MG. However, it is not possible to differentiate both types based only on clinical presentation. Anti-MuSK-positive MG occurs predominately in middle-aged women. Patients with anti-MuSK antibodies may have atypical presentations characterized by prominent oropharyngeal, facial, neck and respiratory muscle weakness. Facial and tongue atrophy may be present by clinical examination and by magnetic resonance imaging (MRI) [ 10 ]. The risk of myasthenic crisis is particularly high, and the chances of achieving complete stable remission are significantly lower than in anti-AChR MG [ 11 ]. As in the anti-AChR antibody-positive MG, neonatal myasthenia occurs in some newborns of myasthenic mothers due to transfer of maternal antibodies [ 12 ]. Furthermore, this type of MG can be also induced by d -penicillamine medication [ 13 ].
Immunological findings suggest that anti-MuSK MG may be different in aetiological and pathological mechanisms from the anti-AChR-positive MG. In anti-AChR MG, antibodies of the immunoglobulin (Ig)G1 and IgG3 subclass modulate the AChR, cause complement activation and attract lymphocytes, acting together to decrease levels of the AChR and AChR-associated proteins and to reduce postsynaptic folding. In patients with anti-MuSK antibodies, there is no evidence of loss of junctional folds and no apparent loss of AChR density. Anti-MuSK antibodies are predominantly of the IgG4 isotype, which differs functionally from other IgG subclasses in its anti-inflammatory activity. The IgG4 antibodies do not cause complement activation at the end-plate. Moreover, IgG4 undergoes a post-translational modification termed ‘Fab arm exchange’ that prevents cross-linking of antigens [ 14 ]. The transfer of immunoglobulins from anti-MuSK-positive MG patients results in end-plate destruction in the recipient animals [ 15 ]. The anti-MuSK antibodies probably interfere with the cellular agrin-MuSK signalling cascade that leads to the formation of the AChR-rapsyn clusters. MuSK also anchors the collagenic tail subunit of acetylcholinesterase, and it was shown recently that passive transfer of anti-MuSK to mice reduces the size and density of ColQ to approximately 10% of controls [ 16 ].
For diagnosis, it is important to notice that needle electromyography (EMG) may show findings suggestive for myopathy in combination with abnormal spontaneous activity. This can easily be misinterpreted as evidence for inflammatory muscle disease [ 17 ]. Single-fibre EMG recordings of limb muscles give often negative results as these muscles are frequently spared and, on account of cholinergic hypersensitivity, many patients do not improve (or even do worse) with cholinesterase inhibitors. Therefore, pharmacological testing with intravenous administration of acetylcholinesterase inhibitors (e.g. edrophonium chloride, the so-called ‘Tensilon test’) is not useful in cases of suspected anti-MuSK MG [ 18 ]. As in the anti-AChR MG, there is a relationship between the individual anti-MuSK serum concentration and disease activity [ 19 ]. In MuSK-MG, thymus is normal or only very slightly affected [ 20 , 21 ]. It seldom shows germinal centres, and those that are present are small and similar to the small germinal centres that can be found in healthy thymus. There is also little or no activation of complement in the thymus. Thus, these observations suggest that the thymus in anti-MuSK myasthenia does not show any of the changes found in anti-AChR myasthenia patients. Considering the lack of evidence, we do not recommend thymectomy to our anti-MuSK MG patients.
Acetylcholinesterase inhibitors, such as pyridostigmine, are often poorly tolerated due to annoying muscle cramps. Intensive immunosuppression is often required. Administration of the anti-CD20 antibody rituximab (see below) often results in a long-lasting, outstandingly good therapeutic effect [ 22 ]. However, controlled prospective study-collected data on the use of rituximab in MG are not yet available. Apheresis and intravenous administration of immunoglobulins are used to overcome severe exacerbations of anti-MuSK MG [ 23 ].
Comorbidities
After confirmation of the diagnosis it is essential to identify comorbidities associated frequently with MG. Due to the possibility of a paraneoplastic aetiology, the search for a thymoma is mandatory. Thymoma-associated MG is generally associated with anti-AChR antibodies and not with anti-MuSK antibodies. In adults younger than 40 years of age, the presence of antibodies against titin or against the recombinant 30-kd titin fragment MGT30, respectively, reliably indicates a thymoma [ 24 ]. Titin is a large protein of the myofibrillar cytoskeleton. It is involved in the maintenance of the sarcomere and binds to the contact point between the A and I bands. Considering the low sensitivity and specificity of serological testing, computerized tomography (CT) or MR imaging studies are mandatory to rule out the presence of a thymoma. The treatment of patients with thymoma does not differ from those patients without thymoma but is complicated by the necessity to treat the neoplasm. Radiotherapy may be useful as adjuvant therapy in cases with incomplete surgical resection with microscopic or macroscopic residual thymoma tissue. Chemotherapy is considered a valid option in selected patients with residual disease after local treatments or to improve resectability of advanced tumours [ 25 , 26 ].
Furthermore, the following conditions are frequently observed in MG patients.
Association with other autoimmune diseases
Other autoimmune diseases are especially thyroiditis, rheumatoid arthritis or lupus erythematosus. Currently, the association of neuromyelitis optica spectrum disorder (NMOSD) with anti-aquaporin-4 antibodies and the anti-AChR antibody-positive MG has been described [ 27 ]. Both immune diseases occur at least 70 times more frequently in common than would be expected by chance. NMOSD develop almost exclusively in females with juvenile or early-onset MG [ 28 ]. MG frequently takes an unusually mild course in patients with NMOSD. A history of thymectomy could be a possible risk factor for the later development of NMOSD. NMOSD symptoms may begin with perennial latency after thymectomy [ 29 ]. In MG patients presenting with atypical motor or optic symptoms, testing for anti-aquaporin-4 antibodies should be ordered [ 28 ].
Inflammatory muscle disorders
In myasthenic patients, the risk of myocarditis is increased [ 30 ]. This association had already been described as ‘Herzmyasthenie’ by Leopold Laquer in 1901. The German term ‘Herzmyasthenie’ means, literally, myasthenia of the heart. Cardiac symptoms, such as shortness of breath and exercise intolerance, might be explained falsely by the existing MG. The possibility of associated myocarditis, especially during exacerbations of MG, must be kept in mind. Of more than 900 Japanese patients, myocarditis was found in three and myositis in six individuals [ 31 ]. Myocarditis developed 13–211 months after the MG onset and was characterized by heart failure and arrhythmias. Myositis, developing before or at the same time as MG, affected limb and paraspinal muscles. Seven patients had one of these autoantibodies to titin, ryanodine receptor and muscular voltage-gated potassium channel Kv1·4, but not myositis-specific autoantibodies. Anti-Kv1·4 antibody-positive patients suffered from severe MG with bulbar symptoms, crisis, thymoma, myocarditis and prolonged QT time on electrocardiography. These results contrast with a study on 129 Caucasian patients [ 32 ]. In this cohort there were 22 (17%) anti-Kv1·4 antibody-positive patients, most of them women with late-onset MG.
Consequences of muscular weakness
Many patients complain of pain because of poor posture caused by the myasthenic weakness. Analgesic medication and orthopaedic therapy, including physiotherapy, may alleviate these frequent complaints. The risk of sleep apnoea syndrome is increased and there is a high prevalence of sleep disturbance among MG patients [ 33 ]. Sleep-related complaints may help to identify subjects at risk for abnormal breathing during sleep, even when daytime functional activity is judged normal.
Treatment options
Treatment of MG is based on four different options which take different amounts of time before muscular weakness will improve ( Fig. 1 ):
improvement of neuromuscular transmission by acetylcholine esterase inhibitors, e.g. pyridostigmine
treatment of acute exacerbations (plasmapheresis, immunoadsorption, intravenous immunoglobulin)
immunosuppression

Onset of action of the different therapy options in myasthenia gravis. The administration of acetylcholinesterase inhibitors (AChE) improves muscular weakness for several hours, but does not affect the course of the disease. At the beginning of corticosteroid therapy there is the risk of deterioration. Long-term therapy with corticosteroid should be avoided. With the use of immunosuppressive drugs, such as azathioprine, an effect on the myasthenic symptoms starts after several months of therapy. Plasma exchange or the intravenous immunoglobulins (IVIg) are used for myasthenic crisis. Both improve myasthenic weakness for just a few weeks. Thymectomy might influence beneficially the long-term course of non-paraneoplastic myasthenia gravis.
For each patient, an individual treatment plan must be compiled that will be adjusted further on the therapeutic response during the course. Using the spectrum of treatment options available nowadays, the majority of myasthenic patients can have largely normal lives. Even complete stable remission can be achieved in nearly every fourth patient suffering from anti-AChR or double-negative MG, respectively [ 11 ]. Unfortunately, treatment response is much less favourable in anti-MuSK MG, with a significantly increased risk of myasthenic crisis [ 34 ]. Experience shows, however, that therapeutic errors occur frequently in MG. For example, the hasty tapering of immunosuppressants is a common mistake resulting in even life-threatening exacerbations of the disease activity. Many myasthenic patients require life-long immunosuppressive therapy.
The current standard treatment of MG is hardly proven by controlled studies. It is largely based on serendipity and retrospective studies. For example, there are no studies on the potential benefit of an early and intensive immunosuppression similar to the ‘hit hard and early’ treatment concept in rheumatoid arthritis. Well-designed clinical trials comparing currently available therapeutic options are lacking. The choice of treatment modalities seems to rely mainly on institutional preferences and the personal experiences of the respective neurologist. However, conducting controlled trials on the therapy of rare and heterogeneous diseases, such as MG, is extremely difficult or even impossible. Moreover, poorly designed studies can even lead to fallacies.
With optimal treatment, the prognosis is good in terms of daily functions, quality of life and survival in the majority of patients suffering from MG. A thorough synopsis of the current standard therapy of MG is beyond the scope of this article. Symptomatic treatment with acetylcholine esterase inhibition is usually combined with immunosuppression. For decades, thymectomy has been performed in younger adults to improve non-paraneoplastic MG [ 35 ]. However, controlled prospective studies on the suspected benefit of this surgical procedure are still lacking. In view of the lack of controlled trials, it is not surprising that neurologists hold differing views to when, and even if, a thymectomy is indicated in the course of non-paraneoplastic MG. Probably the majority of neurologists recommend thymectomy to patients suffering from generalized, early-onset MG and who are positive for anti-AChR antibodies. It is not recommended for anti-MuSK-positive MG (see above). Some neurologists believe that thymectomy is particularly beneficial if it is carried out early in the course of illness. It is unclear if thymectomy may reduce the risk of generalization in patients with purely ocular symptoms. A better understanding of the indications for surgery in non-paraneoplastic MG awaits well-designed prospective studies. In addition, there is no surgical consensus on whether the trans-sternal or the endoscopic procedure should be given preference [ 36 , 37 ]. The final data collection date for primary outcome measure of the MGTX trial will not be announced before August 2015 (ClinicalTrials.gov Identifier: NCT00294658). This international, prospective, randomized trial began in 2006 [ 38 ]. It aims to evaluate the impact of extended transsternal thymectomy on myasthenic symptoms, prednisone requirements and quality of life in patients with non-thymomatous, anti-AChR-positive MG.
Drugs improving neuromuscular transmission: acetylcholinesterase inhibitors
Pyridostigmine bromide is still the most commonly used acetylcholinesterase inhibitor in the treatment of MG, introduced more than 60 years ago [ 39 ]. The administration of acetylcholinesterase inhibitors is symptomatic therapy without affecting the course of the disease with the risk of intoxication at very high daily doses (so-called ‘cholinergic crisis’). The daily dose of pyridostigmine bromide should exceed 600 mg only exceptionally. It is suitable as a long-term treatment in patients with very mild, non-progressive disease, and as an adjunctive therapy in patients with severe disease who are also on immunotherapy. In general, however, pyridostigmine provides only limited benefit in severely affected patients. Pyridostigmine may cause bradycardia, especially in elderly patients. Increased bronchial and oral secretions are a serious problem in patients with swallowing or respiratory insufficiency. Patients on higher daily doses frequently complain about excessive sweating, muscle cramps and diarrhoea. Switching medication to a sustained-release dosage form of pyridostigmine, which is available in only a limited number of countries (e.g. in the United States and Germany), may help to increase tolerability of this medication [ 40 ].
Anti-sense oligonucleotides present a novel type of AChE inhibition. AChE pre-mRNA is susceptible to alternative splicing. MG is associated with expression of the read-through transcript (AChE-R) which, unlike the normal ‘synaptic’ transcript (AChE-S), is not tethered to the post-synaptic membrane, but is a soluble monomer in the synaptic cleft. In experimental autoimmune MG (EAMG), inhibition of production of AChE-R using anti-sense oligonucleotides results in a significant reduction in synaptic expression of AChE-R. This improves muscle strength and increases the survival rates of experimental animals with EAMG [ 41 ]. In humans there are only preliminary data on the therapeutic effect of Monarsen (EN101). This is a synthetic 20-base anti-sense oligodeoxynucleotide directed against the human AChE gene, modified to achieve stability for oral administration. Recent in-vitro and in-vivo studies indicate that EN101 is a Toll-like receptor (TLR)-9-specific ligand that can suppress proinflammatory functions and shift nuclear factor kappa B from the proinflammatory canonical pathway to the anti-inflammatory alternative pathway [ 42 ]. TLR-9 is a member of the TLR family, which plays a fundamental role in pathogen recognition and activation of innate immunity.
Treatment of acute exacerbations
Plasmapheresis, immunoadsorption and the intravenous administration of immunoglobulins, respectively, are used for crisis intervention. Only rarely do patients depend upon one of these therapies for a longer period of time [ 43 ]. Traditional plasma exchange entails removal of the pathogenic antibodies and other plasma components, such as soluble adhesion molecules and cytokines, separation from other blood components and then supplementation with 5% human albumin and crystalloids. The procedure may be carried out by plasma filtration techniques, plasma separation and more recently by antigen-specific immunoadsorption techniques that enable the return of non-pathogenic blood components to the patient. A standard course in MG entails five exchanges on alternating days utilizing 2–4 litres per exchange [ 44 ]. Venous access for plasma exchange can be achieved by central venous catheters or peripheral veins, and the preferred method varies among providers. Very recently, one retrospective study showed that peripheral veins access can be used successfully in most myasthenic patients and reduces the risk of serious and even lethal complications of the procedure [ 45 ].
A number of case reports and smaller, uncontrolled case series showed evidence for a roughly comparable clinical efficacy of plasmapheresis and immunoadsorption. However, the latter method avoids the necessity to substitute plasma replacement solution. This might result in better tolerability. Indeed, the first controlled study comparing the efficacy and safety of both treatments in myasthenic crisis confirms this advantage [ 46 ]. The use of high-dose intravenous immunoglobulin (IVIg) has gained wide application in the treatment of severe MG. Their mechanism of action is quite complex and not fully understood. IVIg seems to affect immune homeostasis by interfering at multiple levels, including modulation of the pathogenic autoantibody response, inhibition of complement activation and interference with the membrane attack complex formation, modulation of Fc receptors, down-regulation of the pathogenic cytokine responses and suppression of T cell function. The procedure usually entails the administration of 0·4 g/kg body weight human pooled IgG over 3 or 5 days [ 44 ]. In acute exacerbations, including myasthenic crisis, intravenous Ig and plasma exchange have good and similar effects [ 47 , 48 ]. The major drawback of both is the relatively short-lived (in general up to 6 weeks) improvement in strength that makes the co-administration of longer acting immunosuppressive or immunomodulatory agents necessary.
Immunosuppressants
Azathioprine still remains the first choice for long-term immunosuppressive therapy. However, it is important to point out that there are only very limited data from controlled studies on the efficacy of azathioprine [ 49 ]. A significant disadvantage of azathioprine is the delayed onset of action. Commonly, azathioprine is therefore started combined with prednisolone to achieve a rapid therapeutic effect. Individually adjusted to the patient's needs, the prednisolone daily dose is then reduced gradually over a prolonged period of time. In a randomized double-blind study of 34 MG patients published in 1998, Palace et al . [ 49 ] compared prednisolone and azathioprine versus prednisolone alone who were followed-up for 3 years. One group received prednisolone (on alternate days) plus azathioprine (2·5 mg/kg); the other group received prednisolone on alternate days plus placebo. Initial high-dose prednisolone (1·5 mg/kg on alternate days) was tapered at remission to the minimal dose required to maintain remission. The prednisolone dose did not differ significantly between the two groups at 1 year but was reduced at 2 and 3 years in the azathioprine group. Patients with refractory disease or azathioprine intolerance are dependent upon alternative corticosteroid sparing immunosuppressive treatment options than azathioprine. Many patients with generalized MG require lifelong immunosuppression.
Corticosteroids are the least expensive, most reliable and rapid-acting drugs for immunomodulation in MG. This appraisal is based on general clinical experience rather than controlled studies. Their long-term use is complicated by severe and often intolerable adverse effects. The optimal manner to initiate corticosteroids depends upon the severity of weakness. It is important to note that with the initiation of higher doses, e.g. 60–100 mg prednisolone per day, some patients develop a significant exacerbation of myasthenic weakness, usually within the first day of treatment. The cause of this clinical phenomenon is elusive. In ocular MG a lower dose of prednisolone for a limited period of time, e.g. 20 mg for 2–4 weeks, can result in a pronounced improvement.
Cyclophosphamide
Cyclophosphamide pulses (500–1000 mg/m 2 ) given every 4–12 weeks has been used occasionally for refractory MG. Alternatively, it can also be given orally in a daily dose of 1–2 mg/kg body weight. Cyclophosphamide may be used only cautiously because of its myelotoxicity. Therefore, it is mandatory to evaluate again the necessity of the cyclophosphamide treatment after 6 months of treatment by omitting the medication or by tapering the daily dose, respectively. In pulse treatment, sufficient diuresis and the adjuvant mesna are required to reduce the risk of haemorrhagic cystitis. Additionally, myocardial damage, pulmonary fibrosis and cancer induction are possible consequences of the use of cyclophosphamide. Drachman et al . [ 50 ] treated three myasthenic patients, for whom treatment with thymectomy, plasmapheresis and conventional immunosuppressive treatment failed, using high-dose cyclophosphamide (50 mg/kg/day intravenously for 4 days) followed by granulocyte colony-stimulating factor. It is known that such immunoablative treatment with high-dose cyclophosphamide does not damage haematopoietic stem cells, permitting repopulation of the immune system without bone marrow transplant. All three patients showed marked improvement in myasthenic weakness.
Methotrexate
Methotrexate is a commonly used alternative to azathioprine. It is an anti-metabolite which has been used for decades in cancer therapy. In low doses, methotrexate is a generally safe and well-tolerated drug in the treatment of certain autoimmune diseases. However, there is only limited evidence for the effectiveness in MG. A recently published, single-blind study from South Africa provides evidence that methotrexate is an effective steroid-sparing agent 10 months after treatment initiation in generalized MG [ 51 ]. This study suggests that methotrexate has similar efficacy and tolerability to azathioprine.
Mycophenolate mofetil
Several retrospective case–series have suggested a very beneficial effect of this immunosuppressant. It is a reversible inhibitor of inosine monophosphate dehydrogenase in purine biosynthesis, which is necessary for T cells and B cells. Other cells are able to recover purines via a separate scavenger pathway and are thus able to escape the effect. Most patients well tolerate daily doses of 1–2·5 g. Two pivotal studies did not confirm the efficacy of mycophenolate in MG [ 52 , 53 ]. However, it is important to note the relevant limitations of both studies. For example, the observation periods of 12 and 36 weeks, respectively, appear to be too short for an assessment of the drug effect in MG. In the open follow-up, patients on mycophenolate monotherapy began to improve between 6 and 12 months. In the combination therapy group, prednisone dose decreased after 12 months [ 54 ]. After more than 24 months, 53% were off prednisone and 75% took less than 7·5 mg prednisone per day. This follow-up corroborates the experience of previous retrospective and pilot studies in demonstrating that mycophenolate is an effective treatment for myasthenic patients as either monotherapy or adjunctive therapy to prednisone. The long follow-up demonstrated a steroid-sparing effect of mycophenolate during the second and third year of therapy that could not be demonstrated by studies of shorter duration. This illustrates that retrospective studies using rigorous outcomes measures can provide valuable information that may not be available from in short-term randomized controlled trials [ 54 ].
Cyclosporin and tacrolimus
Both are powerful immunosuppressive drugs used widely to prevent organ rejection after transplantation and in the treatment of autoimmune diseases. Although not structurally related to cyclosporin, tacrolimus has a similar mechanism of action and it has increasingly replaced cyclosporin for chronic immunosuppression after transplantation. Cyclosporin is a cyclic nonribosomal peptide of 11 amino acids and contains a single d -amino acid. Instead, tacrolimus has a macrolide lactone structure. Both inhibit the transcriptional activation of lymphokine and other genes required for T cell proliferation. The first step in mediating immunosuppressive effects is the binding to their respective intracellular receptors, the immunophilin. The drug/immunophilin complex binds to and inhibits the calcium/calmodulin-dependent serine/threonine phosphatase calcineurin. Under normal circumstances, calcineurin is responsible for activating the transcription of interleukin 2. Possible risks of both drugs include nephrotoxicity, encephalopathy, hypertension and tremor.
Cyclosporin is the immunosuppressive agent that revolutionized organ transplantation in the early 1980s by doubling the 1-year survival rate of cadaveric allografts. However, there are only scattered studies on the use of cyclosporin in MG [ 55–58 ]. In one controlled prospective and placebo-controlled study, 39 patients with steroid-dependent generalized MG received cyclosporin (5 mg/kg per body weight) or placebo for 6 months [ 56 ]. At the end of the study, patients in the cyclosporin group had significantly greater improvement in strength and a reduction in antibody titre. Percentage reduction of steroid medication was greater in the cyclosporin group, although the difference was not statistically significant. Cumulative side effects, however, caused a third of the patients to discontinue the medication; 10% did so secondary to slowly progressive nephrotoxicity. From our point of view, the limited evidence does not justify cyclosporin as a first-line immunosuppressant in MG. It is a reserve drug whose use is restricted due to serious side effects, numerous drug interactions, relatively high therapy costs and the necessity of regular measurements of the cyclosporin blood concentrations.
Tacrolimus suppresses the induction of experimental autoimmune myasthenianin rats [ 59 ]. In several small case–series tacrolimus lowered steroid requirements and induced stable remissions in MG [ 60–63 ]. Recently, a double-blind, placebo-controlled, parallel group study focused on the ability of tacrolimus to reduce the corticosteroid dose in patients with stable myasthenic symptoms on prednisolone at doses equivalent to 10–20 mg/day [ 64 ]. Patients received tacrolimus or placebo for 28 weeks and the corticosteroid dose was tapered with the procedures specified in the study protocol. Unfortunately, this short-duration trial provided a disappointing result regarding the primary efficacy end-point: the two study groups did not differ in the mean daily steroid dose. Besides immunosuppression, tacrolimus might have the potential to increase muscle strength by enhancing this ryanodine receptor function [ 65 ]. However, it is doubtful if this effect is indeed of clinical significance.
Emerging therapy options
The use of monoclonal antibodies with an innovative mode of action is promising, and might change the treatment of MG significantly in the coming years. A particularly promising candidate is rituximab. This is a genetically engineered chimeric mouse/human monoclonal antibody representing a glycosylated immunoglobulin with human IgG1 constant regions and murine light- and heavy-chain variable region sequences [ 66–69 ]. The antibody is produced by mammalian (Chinese hamster ovary) cell suspension culture. It is approved for the treatment of some lymphoma types and of severe active rheumatoid arthritis in adults who have had an inadequate response or intolerance to other disease modifying anti-rheumatic drugs. Rituximab binds specifically to the transmembrane antigen, CD20, a non-glycosylated phosphoprotein, located on pre-B and mature B lymphocytes. CD20 is found on both normal and malignant B cells, but not on haematopoietic stem cells, pro-B cells, normal plasma cells or other normal tissue. Toxicity studies have shown no other effect than the expected pharmacological depletion of B cells in peripheral blood and in lymphoid tissue. Peripheral B cell counts decline below normal following completion of the first dose of rituximab. B cell repletion begins within 6 months of treatment returning to normal levels between 9 and 12 months after completion of therapy [ 66 , 67 ].
Rituximab provides promising expectations for the treatment of MG, although no randomized controlled trials have been conducted so far. Case reports, retrospective small series and uncontrolled studies describe a long-lasting and pronounced clinical benefit due to rituximab even in severely affected, drug-resistant patients with MG [ 22 , 70–78 ]. Most interestingly, this might give better clinical benefit and last longer in anti-MuSK than in anti-AChR MG patients. Diaz-Manera et al . [22] treated six anti-MuSK- and 11 anti-AChR-positive MG patients with rituximab. All the patients included in this study were resistant to previous therapies and were classes III to V in the Myasthenia Gravis Foundation America classification. MuSK antibodies decreased dramatically during the follow-up after a single rituximab cycle. However, AChR antibodies remained at the same titres during the same period of time. After a mean post-treatment period of 31 months, 10 of the anti-AChR patients improved but six of them needed reinfusions. In contrast, all anti-MuSK patients achieved a remission or minimal manifestations status and no reinfusions were needed. Consequently, in the anti-MuSK group, prednisone doses were reduced significantly and concomitant immunosuppressants could be withdrawn.
Infusion reactions including fever, chills and shivering are the most common side effects of rituximab. Pretreatment with hydrocortisone and diphenhydramine ameliorates these reactions. Very rare cases of progressive multi-focal leucoencephalopathy (PML) have been reported during the use of rituximab in lymphoma patients. The majority of patients suffering from this life-threatening complication had received rituximab in combination with chemotherapy. Cases of fatal PML have been reported following off-label use of rituximab for the treatment of certain autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis and vasculitis. These patients with autoimmune diseases had a history of prior or concurrent immunosuppressive therapy and were diagnosed with PML within 12 months of their last infusion of rituximab [ 79–82 ]. To date, there are no reports on the occurrence of PML in patients treated with rituximab for MG.
This is a proteasome inhibitor approved for treating patients with multiple myeloma, a plasma cell malignancy. Recent preclinical studies in cell cultures and animal models, and clinical studies in organ-transplant recipients, have demonstrated that bortezomib can kill non-neoplastic plasma cells within hours. This suggests that proteasome inhibitors could also be used for rapidly reducing autoantibody production in MG [ 83 ].
B cell activating factor (BAFF) is a potent B cell survival factor, and plays an essential role in B cell homeostasis and B cell function in the periphery [ 84 , 85 ]. Because BAFF is a crucial and potent factor for the survival and growth of B cells, both normal and autoreactive B cells compete for available BAFF. BAFF levels appear to regulate the survival threshold for B cells. Autoreactive B cells are poorly competitive for survival and they appear to be more dependent upon BAFF for their survival. BAFF levels are increased in the circulation of MG patients [ 86 ]. BAFF antagonists such as the monoclonal antibody belimumab may well provide new treatment options for MG. Belimumab is already approved in the United States, Canada and Europe for treatment of systemic lupus erythematosus.
Complement activation at the neuromuscular junction may be the primary cause of acetylcholine receptor loss and failure of neuromuscular transmission seen in MG. Eculizumab, a humanized monoclonal antibody, blocks the formation of terminal complement complex by selectively preventing the enzymatic cleavage of C5. It is the first therapy approved for the treatment of paroxysmal nocturnal haemoglobinuria. The results of one pilot Phase II study on the use of eculizumab in severe and refractory generalized MG are published in abstract form [ 87 ]. The purpose of this study was to determine whether or not eculizumab is safe and effective, despite treatment with various immunosuppressants that are currently available. The study has shown a significant clinical benefit of eculizumab in improving MG compared to placebo. Eculizumab was safe and well tolerated in all treated patients.
Etanercept blocks tumour necrosis factor (TNF)-α activity. It has European approval to treat rheumatoid arthritis, juvenile rheumatoid arthritis and psoriatic arthritis, plaque psoriasis and ankylosing spondylitis. In animals, it suppresses ongoing experimental MG [ 88 ]. This observation resulted in a small prospective trial on the effect in corticosteroid-dependent MG. Eleven patients were enrolled, with eight completing this 6-month trial. Two patients were withdrawn owing to disease worsening, and one patient was withdrawn because of an erythematous skin rash. Six of the eight patients who completed the trial improved, based on quantitative measures of muscle strength and lowering of corticosteroid requirement [ 89 ]. In addition to these disappointing study results, there are scattered case reports on patients who developed MG while taking etanercept and had resolution of symptoms after stopping it [ 90 ].
Granulocyte–macrophage colony-stimulating factor (GM-CSF)
Forkhead box protein 3 (FoxP3) is a transcription factor necessary for the function of regulatory T cells (T regs ) [ 91 ]. T regs maintain immune homeostasis and self-tolerance and play an important role in the prevention of autoimmune disease. In-vitro administration of GM-CSF enhances the suppressive function of T regs and up-regulated FoxP3 expression in T regs . There is one single case report on a patient with a prolonged myasthenic crisis refractory to conventional immunomodulatory treatments who was treated with GM-CSF [ 92 ]. This 77-year-old man received 750 μg GM-CSF daily for 3 days followed by 250 μg/day for 3 days. After the fifth dose of GM-CSF, he had improved generalized strength and was eventually weaned from the ventilator.
The pillars of current MG therapy were already introduced more than 40 years ago. These are still pyridostigmine, corticosteroids, azathioprine and thymectomy. However, first experiences with new drugs, e.g. rituximab, are highly promising. It could be that the treatment of MG will change substantially until the end of this decade, but we are still far from a targeted immunotherapy.
The author discloses no conflicts of interest.
Pakzad Z , Aziz T , Oger J . Increasing incidence of myasthenia gravis among elderly in British Columbia, Canada . Neurology 2011 ; 76 : 1526 – 1528 .
Google Scholar
Pallaver F , Riviera AP , Piffer S et al. Change in myasthenia gravis epidemiology in Trento, Italy, after twenty years . Neuroepidemiology 2011 ; 36 : 282 – 287 .
Matsuda M , Dohi-Iijima N , Nakamura A et al. Increase in incidence of elderly-onset patients with myasthenia gravis in Nagano Prefecture, Japan . Intern Med 2005 ; 44 : 572 – 577 .
Zivkovic SA , Clemens PR , Lacomis D . Characteristics of late-onset myasthenia gravis . J Neurol 2012 ; 259 : 2167 – 2171 .
Leite MI , Jacob S , Viegas S et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis . Brain 2008 ; 131 : 1940 – 1952 .
Wu H , Xiong WC , Mei L . To build a synapse: signaling pathways in neuromuscular junction assembly . Development 2010 ; 137 : 1017 – 1033 .
Hoch W , McConville J , Helms S , Newsom-Davis J , Melms A , Vincent A . Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies . Nat Med 2001 ; 7 : 365 – 368 .
Pevzner A , Schoser B , Peters K et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis . J Neurol 2012 ; 259 : 427 – 435 .
Zhang B , Tzartos JS , Belimezi M et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis . Arch Neurol 2012 ; 69 : 445 – 451 .
Farrugia ME , Robson MD , Clover L et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis . Brain 2006 ; 129 : 1481 – 1492 .
Baggi F , Andreetta F , Maggi L et al. Complete stable remission and autoantibody specificity in myasthenia gravis . Neurology 2013 ; 80 : 188 – 195 .
Behin A , Mayer M , Kassis-Makhoul B et al. Severe neonatal myasthenia due to maternal anti-MuSK antibodies . Neuromuscul Disord 2008 ; 18 : 443 – 446 .
Poulas K , Koutsouraki E , Kordas G , Kokla A , Tzartos SJ . Anti-MuSK- and anti-AChR-positive myasthenia gravis induced by d-penicillamine . J Neuroimmunol 2012 ; 250 : 94 – 98 .
Gomez AM , Van Den Broeck J , Vrolix K et al. Antibody effector mechanisms in myasthenia gravis-pathogenesis at the neuromuscular junction . Autoimmunity 2010 ; 43 : 353 – 370 .
Cole RN , Reddel SW , Gervasio OL , Phillips WD . Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction . Ann Neurol 2008 ; 63 : 782 – 789 .
Kawakami Y , Ito M , Hirayama M et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK . Neurology 2011 ; 77 : 1819 – 1826 .
Guptill JT , Sanders DB , Evoli A . Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts . Muscle Nerve 2011 ; 44 : 36 – 40 .
Pasnoor M , Wolfe GI , Nations S et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a U.S. experience . Muscle Nerve 2010 ; 41 : 370 – 374 .
Niks EH , van Leeuwen Y , Leite MI et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1 . J Neuroimmunol 2008 ; 195 : 151 – 156 .
Lauriola L , Ranelletti F , Maggiano N et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis . Neurology 2005 ; 64 : 536 – 538 .
Leite MI , Strobel P , Jones M et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG . Ann Neurol 2005 ; 57 : 444 – 448 .
Diaz-Manera J , Martinez-Hernandez E , Querol L et al. Long-lasting treatment effect of rituximab in MuSK myasthenia . Neurology 2012 ; 78 : 189 – 193 .
Takahashi H , Kawaguchi N , Nemoto Y , Hattori T . High-dose intravenous immunoglobulin for the treatment of MuSK antibody-positive seronegative myasthenia gravis . J Neurol Sci 2006 ; 247 : 239 – 241 .
Voltz RD , Albrich WC , Nagele A et al. Paraneoplastic myasthenia gravis: detection of anti-MGT30 (titin) antibodies predicts thymic epithelial tumor . Neurology 1997 ; 49 : 1454 – 1457 .
Spaggiari L , Casiraghi M , Guarize J . Multidisciplinary treatment of malignant thymoma . Curr Opin Oncol 2012 ; 24 : 117 – 122 .
Venuta F , Rendina EA , Anile M , de Giacomo T , Vitolo D , Coloni GF . Thymoma and thymic carcinoma . Gen Thorac Cardiovasc Surg 2012 ; 60 : 1 – 12 .
Leite MI , Coutinho E , Lana-Peixoto M et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients . Neurology 2012 ; 78 : 1601 – 1607 .
Jarius S , Paul F , Franciotta D et al. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature . Mult Scler 2012 ; 18 : 1135 – 1143 .
Kister I , Gulati S , Boz C et al. Neuromyelitis optica in patients with myasthenia gravis who underwent thymectomy . Arch Neurol 2006 ; 63 : 851 – 856 .
Aarli JA . Herzmyasthenie: myasthenia of the heart . Arch Neurol 2009 ; 66 : 1322 – 1323 .
Suzuki S , Utsugisawa K , Yoshikawa H et al. Autoimmune targets of heart and skeletal muscles in myasthenia gravis . Arch Neurol 2009 ; 66 : 1334 – 1338 .
Romi F , Suzuki S , Suzuki N , Petzold A , Plant GT , Gilhus NE . Anti-voltage-gated potassium channel Kv1.4 antibodies in myasthenia gravis . J Neurol 2012 ; 259 : 1312 – 1316 .
De Lapiscina EH , Aguirre ME , Blanco TA , Pascual IJ . Myasthenia gravis: sleep quality, quality of life, and disease severity . Muscle Nerve 2012 ; 46 : 174 – 180 .
Deymeer F , Gungor-Tuncer O , Yilmaz V et al. Clinical comparison of anti-MuSK- vs anti-AChR-positive and seronegative myasthenia gravis . Neurology 2007 ; 68 : 609 – 611 .
Sonett JR , Jaretzki A , III . Thymectomy for nonthymomatous myasthenia gravis: a critical analysis . Ann NY Acad Sci 2008 ; 1132 : 315 – 328 .
Youssef SJ , Louie BE , Farivar AS , Blitz M , Aye RW , Vallieres E . Comparison of open and minimally invasive thymectomies at a single institution . Am J Surg 2010 ; 199 : 589 – 593 .
Lin MW , Chang YL , Huang PM , Lee YC . Thymectomy for non-thymomatous myasthenia gravis: a comparison of surgical methods and analysis of prognostic factors . Eur J Cardiothorac Surg 2010 ; 37 : 7 – 12 .
Newsom-Davis J , Cutter G , Wolfe GI et al. Status of the thymectomy trial for nonthymomatous myasthenia gravis patients receiving prednisone . Ann NY Acad Sci 2008 ; 1132 : 344 – 347 .
Maggi L , Mantegazza R . Treatment of myasthenia gravis: focus on pyridostigmine . Clin Drug Invest 2011 ; 31 : 691 – 701 .
Sieb JP , Köhler W . Benefits from sustained-release pyridostigmine bromide in myasthenia gravis: results of a prospective multicenter open-label trial . Clin Neurol Neurosurg 2010 ; 112 : 781 – 784 .
Sussman JD , Argov Z , McKee D , Hazum E , Brawer S , Soreq H . Antisense treatment for myasthenia gravis: experience with monarsen . Ann NY Acad Sci 2008 ; 1132 : 283 – 290 .
Sussman J , Argov Z , Wirguin Y , Apolski S , Milic-Rasic V , Soreq H . Further developments with antisense treatment for myasthenia gravis . Ann NY Acad Sci 2012 ; 1275 : 13 – 16 .
Wagner S , Janzen RW , Mohs C , Pohlmann S , Klingel R , Grützmacher PW . Long-term treatment of refractory myasthenia gravis with immunoadsorption . Dtsch Med Wochenschr 2008 ; 133 : 2377 – 2382 [in German].
Jani-Acsadi A , Lisak RP . Myasthenic crisis: guidelines for prevention and treatment . J Neurol Sci 2007 ; 261 : 127 – 133 .
Guptill JT , Oakley D , Kuchibhatla M et al. A Retrospective study of complications of therapeutic plasma exchange in myasthenia . Muscle Nerve 2013 ; 47 : 170 – 176 .
Köhler W , Bucka C , Klingel R . A randomized and controlled study comparing immunoadsorption and plasma exchange in myasthenic crisis . J Clin Apher 2011 ; 26 : 347 – 355 .
Barth D , Nabavi NM , Ng E , Nwe P , Bril V . Comparison of IVIg and PLEX in patients with myasthenia gravis . Neurology 2011 ; 76 : 2017 – 2023 .
Mandawat A , Kaminski HJ , Cutter G , Katirji B , Alshekhlee A . Comparative analysis of therapeutic options used for myasthenia gravis . Ann Neurol 2010 ; 68 : 797 – 805 .
Palace J , Newsom-Davis J , Lecky B , Myasthenia Gravis Study Group . A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis . Neurology 1998 ; 50 : 1778 – 1783 .
Drachman DB , Jones RJ , Brodsky RA . Treatment of refractory myasthenia: ‘rebooting’ with high-dose cyclophosphamide . Ann Neurol 2003 ; 53 : 29 – 34 .
Heckmann JM , Rawoot A , Bateman K , Renison R , Badri M . A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis . BMC Neurol 2011 ; 11 : 97 .
Sanders DB , Hart IK , Mantegazza R et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis . Neurology 2008 ; 71 : 400 – 406 .
Muscle Study Group . A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis . Neurology 2008 ; 71 : 394 – 399 .
Hehir MK , Burns TM , Alpers J , Conaway MR , Sawa M , Sanders DB . Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients . Muscle Nerve 2010 ; 41 : 593 – 598 .
Schalke B , Kappos L , Rohrbach E et al. Cyclosporin A versus azathioprine in the treatment of myasthenia gravis: final results of a randomized, controlled double-blind clinical trial . Neurology 1988 ; 38 ( Suppl. 1 ): 135 .
Tindall RS , Phillips JT , Rollins JA , Wells L , Hall K . A clinical therapeutic trial of cyclosporine in myasthenia gravis . Ann NY Acad Sci 1993 ; 681 : 539 – 551 .
Bonifati DM , Angelini C . Long-term cyclosporine treatment in a group of severe myasthenia gravis patients . J Neurol 1997 ; 244 : 542 – 547 .
Nagane Y , Suzuki S , Suzuki N , Utsugisawa K . Two-year treatment with cyclosporine microemulsion for responder myasthenia gravis patients . Eur Neurol 2010 ; 64 : 186 – 190 .
Yoshikawa H , Iwasa K , Satoh K , Takamori M . FK506 prevents induction of rat experimental autoimmune myasthenia gravis . J Autoimmun 1997 ; 10 : 11 – 16 .
Ponseti JM , Gamez J , Azem J et al. Post-thymectomy combined treatment of prednisone and tacrolimus versus prednisone alone for consolidation of complete stable remission in patients with myasthenia gravis: a non-randomized, non-controlled study . Curr Med Res Opin 2007 ; 23 : 1269 – 1278 .
Evoli A , Di SC , Marsili F , Punzi C . Successful treatment of myasthenia gravis with tacrolimus . Muscle Nerve 2002 ; 25 : 111 – 114 .
Ponseti JM , Fort JM , Espin E , Armengol M . Tacrolimus (FK506) in the treatment of prednisone-resistant myasthenia gravis. Preliminary results of 20 cases . Med Clin (Barc) 2002 ; 118 : 117 [In Spanish].
Minami N , Fujiki N , Doi S et al. Five-year follow-up with low-dose tacrolimus in patients with myasthenia gravis . J Neurol Sci 2011 ; 300 : 59 – 62 .
Yoshikawa H , Kiuchi T , Saida T , Takamori M . Randomised, double-blind, placebo-controlled study of tacrolimus in myasthenia gravis . J Neurol Neurosurg Psychiatry 2011 ; 82 : 970 – 977 .
Imai T , Tsuda E , Hozuki T et al. Early effect of tacrolimus in improving excitation–contraction coupling in myasthenia gravis . Clin Neurophysiol 2012 ; 123 : 1886 – 1890 .
Onrust SV , Lamb HM , Balfour JA . Rituximab . Drugs 1999 ; 58 : 79 – 88 .
Cheson BD . Rituximab: clinical development and future directions . Expert Opin Biol Ther 2002 ; 2 : 97 – 110 .
Rommer PS , Zettl UK , Kieseier B . Requirement for safety monitoring for approved multiple sclerosis therapies: an overview . Clin Exp Immunol 2014 ; 175 : 397 – 407 .
Rommer PS , Dudesek A , Stüve O and Zettl UK . Monoclonal antibodies in treatment of multiple sclerosis . Clin Exp Immunol 2014 ; 175 : 373 – 384 .
Collongues N , Casez O , Lacour A et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study . Muscle Nerve 2012 ; 46 : 687 – 691 .
Stieglbauer K , Topakian R , Schäffer V , Aichner FT . Rituximab for myasthenia gravis: three case reports and review of the literature . J Neurol Sci 2009 ; 280 : 120 – 122 .
Stein B , Bird SJ . Rituximab in the treatment of MuSK antibody-positive myasthenia gravis . J Clin Neuromuscul Dis 2011 ; 12 : 163 – 164 .
Nelson RP Jr , Pascuzzi RM , Kessler K et al. Rituximab for the treatment of thymoma-associated and de novo myasthenia gravis: 3 cases and review . J Clin Neuromuscul Dis 2009 ; 10 : 170 – 177 .
Maddison P , McConville J , Farrugia ME et al. The use of rituximab in myasthenia gravis and Lambert–Eaton myasthenic syndrome . J Neurol Neurosurg Psychiatry 2011 ; 82 : 671 – 673 .
Zebardast N , Patwa HS , Novella SP , Goldstein JM . Rituximab in the management of refractory myasthenia gravis . Muscle Nerve 2010 ; 41 : 375 – 378 .
Lindberg C , Bokarewa M . Rituximab for severe myasthenia gravis – experience from five patients . Acta Neurol Scand 2010 ; 122 : 225 – 228 .
Evoli A , Bianchi MR , Riso R et al. Response to therapy in myasthenia gravis with anti-MuSK antibodies . Ann NY Acad Sci 2008 ; 1132 : 76 – 83 .
Kerkeni S , Marotte H , Miossec P . Improvement with rituximab in a patient with both rheumatoid arthritis and myasthenia gravis . Muscle Nerve 2008 ; 38 : 1343 – 1345 .
Molloy ES . PML and rheumatology: the contribution of disease and drugs . Cleve Clin J Med 2011 ; 78 ( Suppl. 2 ): S28 – S32 .
Bharat A , Xie F , Baddley JW et al. Incidence and risk factors for progressive multifocal leukoencephalopathy among patients with selected rheumatic diseases . Arthritis Care Res (Hoboken) 2012 ; 64 : 612 – 615 .
Palazzo E , Yahia SA . Progressive multifocal leukoencephalopathy in autoimmune diseases . Joint Bone Spine 2012 ; 79 : 351 – 355 .
Molloy ES , Calabrese LH . Progressive multifocal leukoencephalopathy associated with immunosuppressive therapy in rheumatic diseases: evolving role of biologic therapies . Arthritis Rheum 2012 ; 64 : 3043 – 3051 .
Gomez AM , Willcox N , Molenaar PC et al. Targeting plasma cells with proteasome inhibitors: possible roles in treating myasthenia gravis? Ann NY Acad Sci 2012 ; 1274 : 48 – 59 .
Ragheb S , Lisak RP . B-cell-activating factor and autoimmune myasthenia gravis . Autoimmune Dis 2011 ; 2011 : 939520 .
Lisak RP , Ragheb S . The role of B cell-activating factor in autoimmune myasthenia gravis . Ann NY Acad Sci 2012 ; 1274 : 60 – 67 .
Ragheb S , Lisak R , Lewis R , Van SG , Gonzales F , Simon K . A potential role for B-cell activating factor in the pathogenesis of autoimmune myasthenia gravis . Arch Neurol 2008 ; 65 : 1358 – 1362 .
Howard JF Jr , Barohn RJ , Cutter GR et al. A randomized, double-blind, placebo-controlled Phase II study of eculizumab in patients with refractory generalized myasthenia gravis . Muscle Nerve 2013 ; 48 : 76 – 84 .
Christadoss P , Goluszko E . Treatment of experimental autoimmune myasthenia gravis with recombinant human tumor necrosis factor receptor Fc protein . J Neuroimmunol 2002 ; 122 : 186 – 190 .
Rowin J , Meriggioli MN , Tuzun E , Leurgans S , Christadoss P . Etanercept treatment in corticosteroid-dependent myasthenia gravis . Neurology 2004 ; 63 : 2390 – 2392 .
Fee DB , Kasarskis EJ . Myasthenia gravis associated with etanercept therapy . Muscle Nerve 2009 ; 39 : 866 – 870 .
Thiruppathi M , Rowin J , Li JQ , Sheng JR , Prabhakar BS , Meriggioli MN . Functional defect in regulatory T cells in myasthenia gravis . Ann NY Acad Sci 2012 ; 1274 : 68 – 76 .
Rowin J , Thiruppathi M , Arhebamen E , Sheng J , Prabhakar BS , Meriggioli MN . Granulocyte macrophage colony-stimulating factor treatment of a patient in myasthenic crisis: effects on regulatory T cells . Muscle Nerve 2012 ; 46 : 449 – 453 .
Email alerts
Citing articles via.
- About Clinical & Experimental Immunology
- Recommend to Your Librarian
- Advertising and Corporate Services
- Journals Career Network
Affiliations
- Online ISSN 1365-2249
- Copyright © 2024 British Society for Immunology
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
- Open access
- Published: 06 November 2007
Myasthenia gravis
- Vern C Juel 1 &
- Janice M Massey 1
Orphanet Journal of Rare Diseases volume 2 , Article number: 44 ( 2007 ) Cite this article
67k Accesses
126 Citations
10 Altmetric
Metrics details
Myasthenia gravis (MG) is a rare, autoimmune neuromuscular junction disorder. Contemporary prevalence rates approach 1/5,000. MG presents with painless, fluctuating, fatigable weakness involving specific muscle groups. Ocular weakness with asymmetric ptosis and binocular diplopia is the most typical initial presentation, while early or isolated oropharyngeal or limb weakness is less common. The course is variable, and most patients with initial ocular weakness develop bulbar or limb weakness within three years of initial symptom onset. MG results from antibody-mediated, T cell-dependent immunologic attack on the endplate region of the postsynaptic membrane. In patients with fatigable muscle weakness, the diagnosis of MG is supported by: 1. pharmacologic testing with edrophonium chloride that elicits unequivocal improvement in strength; 2. electrophysiologic testing with repetitive nerve stimulation (RNS) studies and/or single-fiber electromyography (SFEMG) that demonstrates a primary postsynaptic neuromuscular junctional disorder; and 3. serologic demonstration of acetylcholine receptor (AChR) or muscle-specific tyrosine kinase (MuSK) antibodies. Differential diagnosis includes congenital myasthenic syndromes, Lambert Eaton syndrome, botulism, organophosphate intoxication, mitochondrial disorders involving progressive external ophthalmoplegia, acute inflammatory demyelinating polyradiculoneuropathy (AIDP), motor neuron disease, and brainstem ischemia. Treatment must be individualized, and may include symptomatic treatment with cholinesterase inhibitors and immune modulation with corticosteroids, azathioprine, cyclosporine, and mycophenolate mofetil. Rapid, temporary improvement may be achieved for myasthenic crises and exacerbations with plasma exchange (PEX) or intravenous immunoglobulin (IVIg). Owing to improved diagnostic testing, immunotherapy, and intensive care, the contemporary prognosis is favorable with less than five percent mortality and nearly normal life expectancy.
Disease name
Myasthenia gravis, Autoimmune myasthenia gravis
Included diseases
Autoimmune myasthenia gravis (MG) encompasses all of the immunologically-mediated disorders affecting the endplate region of the postsynaptic neuromuscular junction. Nearly all of these disorders involve a loss of immunological self-tolerance, though transitory neonatal MG is a self-limited disorder that follows passive transfer of maternal antibodies to the fetus. Congenital myasthenic syndromes stem from genetic mutations that result in abnormal neuromuscular transmission.
MG is termed ocular MG when weakness is exclusive to the eyelids and extraocular muscles, and generalized MG when weakness extends beyond these ocular muscles. Seropositive (SP) MG defines disease with circulating antibodies to the acetylcholine receptor (AChR), while seronegative (SN) patients lack these antibodies. Recently, antibodies to muscle-specific tyrosine kinase (MuSK) have been demonstrated in over 40% of patients with generalized, SN MG [ 1 – 5 ].
Definition and diagnostic criteria
MG remains one of the most challenging medical diagnoses due to its fluctuating character and to the similarity of its symptoms to those of other disorders. Although a formal clinical classification system and research standards have been established for MG, [ 6 ] there are no widely accepted formal diagnostic criteria. The most important elements of diagnosis are clinical history and examination findings of fluctuating and fatigable weakness, particularly involving extraocular and bulbar muscles. A clinical diagnosis may be confirmed by laboratory testing including: 1. pharmacologic testing with edrophonium chloride that elicits unequivocal improvement in strength; 2. electrophysiologic testing with repetitive nerve stimulation (RNS) studies and/or single-fiber electromyography (SFEMG) that demonstrates a primary postsynaptic neuromuscular junctional disorder; or 3. by serological demonstration of AChR or MuSK antibodies.
Epidemiology
Although MG is rare, prevalence rates for MG have increased over time, likely due to improvements in diagnosis and treatment. Recent prevalence rates approach 20/100,000 [ 7 ]. A wide range of incidence is reported with an estimate of about 2.0 to 10.4/million/year in Virginia [ 8 ] to 21.27/million/year in Barcelona, Spain [ 9 ]. The onset of MG is influenced by gender and age in a bimodal fashion. In patients younger than 40, women predominate with a ratio of 7:3. In the fifth decade, new cases of MG are evenly distributed between men and women. After age 50, new cases of MG are slightly more common in men with a ratio of 3:2 [ 10 , 11 ].
Pediatric MG is very rare. Juvenile MG is an autoimmune disorder, while congenital MG results from genetic mutations that impair neuromuscular transmission. Transient neonatal MG is a self-limited disorder related to placental antibody transfer in maternal autoimmune MG. It may be difficult to make the distinction between juvenile MG and congenital MG, particularly in the absence of AChR or MuSK antibodies, or a clear history of ptosis and other manifestations of hypotonia from the time of birth that would suggest genetic disease. These issues are discussed in depth by Andrews [ 12 ].
Clinical description
In MG, patients present with fluctuating and fatigable weakness of specific muscle groups rather than with generalized fatigue or pain. The weakness is variable from day to day and from hour to hour, but it is generally worse later in the day. Sustained exercise and increased body temperature may increase the degree of weakness. Ocular weakness with asymmetric ptosis and binocular diplopia is the most common initial presentation, while early or isolated oropharyngeal or limb weakness is less common.
Ocular weakness presents as fluctuating, fatigable, and sometimes alternating ptosis and binocular diplopia that resolves with closing or covering one eye. Many patients report difficulties with driving, reading, or watching television. Bright lights may be quite bothersome. Retrospectively, many patients report periods of intermittent blurred vision before they were able to discern dual visual images. Examination may demonstrate asymmetrical weakness of multiple extraocular muscles that cannot be attributed to a single cranial neuropathy. Pupillary function is normal. Ptosis may be elicited or increased with sustained upgaze. In MG, ptosis is generally asymmetrical, and it may be associated with ipsilateral frontalis muscle contraction to help compensate for the weak levator palpebrae. Excessive lid elevation or Cogan's lid twitch sign may be observed when gaze is directed from down to upward.
Dysthyroid (Graves) ophthalmopathy may coexist with ocular MG. Although external ophthalmoplegia may occur in either disorder, dysthyroid ophthalmopathy produces proptosis, not ptosis, owing to enlarged extraocular muscles. The enlarged muscles may be demonstrated by orbital magnetic resonance imaging (MRI).
Jaw closure muscles are frequently affected in MG, but strength is usually normal in jaw opening muscles. Patients may complain of difficulty in chewing candy or tough meats, and some modify their diet to compensate for this difficulty. Some patients assume a thoughtful resting posture by placing the thumb beneath the chin in order to hold the jaw closed. The jaw closure muscles can be examined by exerting several seconds of sustained downward pressure on the chin while the patient attempts to hold the jaw closed.
Many patients exhibit a depressed or expressionless facial appearance. Actions such as whistling, using straws, or inflating balloons may be impaired. A "myasthenic snarl" may be observed when the patient attempts to smile. The snarl follows contraction of the middle portion of the upper lip while the upper mouth corners fail to contract. On examination, many patients exhibit weak forced eye closure that can easily be overcome by the examiner. Bell's phenomenon with upward and lateral rotation of the eyes on attempted closure is observed when the examiner defeats a patient's forced eye closure. Patients with mild lower facial weakness develop a transverse pucker when they attempt to hold air within inflated cheeks. With more overt lower facial weakness, air readily escapes through the lips when the cheeks are squeezed. In severe lower facial weakness, the lips cannot be voluntarily opposed.
Oropharyngeal muscle weakness produces dysarthria and dysphagia. With weakness of palatal muscles, nasal speech develops as air escapes through the nose. This may become increasingly apparent with prolonged speaking. Liquid may also escape through the nose during attempted swallowing with nasal regurgitation. Myasthenic weakness of laryngeal muscles is associated with a hoarse, breathy voice. Incomplete glottic closure during swallowing may produce aspiration. Examination may reveal reduced or absent palate elevation. Tongue weakness may be demonstrated when the patient attempts to protrude either cheek with the tongue against the resistance of the examiner's finger applied to the cheek. With marked tongue weakness, the patient may be unable to protrude the cheek in the absence of applied resistance by the examiner. With severe lingual weakness, the tongue may not protrude beyond the lips. When myasthenic tongue weakness is chronic, tongue atrophy and triple furrowing may develop with accentuated median and lateral lingual furrows.
Neck flexor and extensor muscles are often weak in MG. Though the neck flexors are usually weaker, a "dropped head syndrome" due to neck extensor muscle weakness may occur. Although painless weakness is the rule in MG, patients with neck extensor weakness may experience posterior neck myalgias.
Limb weakness in MG may be associated with complaints of difficulty performing overhead tasks with the arms, and there may be difficulty climbing stairs due to lower extremity weakness. Examination reveals asymmetrical weakness involving any muscle group in the limbs, though the deltoids, triceps brachii, wrist and finger extensors, and foot dorsiflexors are often involved.
Autoimmune MG results from antibody-mediated, T cell-dependent immunologic attack on the postsynaptic membrane of the neuromuscular junction. Abnormal neuromuscular transmission and clinical weakness in MG result from the effects of antibodies that bind to various epitopes of the skeletal muscle endplate region. In most cases, antibodies bind to the main immunogenic region of the α-subunit of the AChR, though MG patients with antibodies to MuSK exhibit clinical weakness and electrophysiologic findings that are quite similar to MG patients with AChR antibodies. MuSK initiates aggregation of AChRs at the muscle endplate during development, [ 2 ] but the function of MuSK in mature skeletal muscle and the pathophysiology of MG related to MuSK antibodies are currently unknown.
In SP MG, binding of antibody to the AChR initiates autoimmune attack on the endplate region. Subsequent damage to the postsynaptic membrane results in simplification of the normal, highly-infolded surface that is accompanied by reduced number and density of AChR [ 13 ]. The functional loss of AChRs reduces the probability of successful neuromuscular transmission following quantal release of acetylcholine by the motor nerve terminal, resulting in clinical weakness in striated muscles.
MG and other autoimmune disorders result from the loss of tolerance to self-antigens. T-lymphocyte tolerance to self-antigens is established in the thymus, and thymic abnormalities are often present in MG. Thymic hyperplasia is observed in about 65% of MG patients, and thymomas are present in about 10% of MG patients [ 14 ]. MG patients with thymoma have more severe and generalized weakness, higher AChR antibody titers, and more severe electrophysiologic abnormalities. Accordingly, patients with SN and ocular MG are less likely to have thymomas. Most thymic tumors are benign, well differentiated, and encapsulated. Patients with MG should undergo chest computed tomography (CT) to exclude the presence of thymoma. While thymoma resection is necessary to prevent compromise of mediastinal structures, the benefit of thymectomy for patients with non-thymomatous MG remains uncertain.
Diagnostic methods
Pharmacologic testing, edrophonium testing.
Edrophonium chloride is an acetylcholinesterase inhibitor with rapid onset (approximately 30 seconds) and short duration (approximately 5 minutes) of pharmacologic action. Edrophonium chloride temporarily improves the safety factor of neuromuscular transmission and may elicit improved strength in patients with abnormal neuromuscular transmission. Edrophonium testing is considered positive when unequivocal improvement in strength follows intravenous administration of edrophonium. Development of increased weakness may also suggest abnormal neuromuscular transmission. The primary limitation of edrophonium testing relates to selection of an objective muscle strength parameter for assessment. Therefore, edrophonium testing is most useful in patients who have significant ptosis or restricted extraocular movements that can be graded objectively. In other muscles, volition and the muscarinic effects of edrophonium may complicate strength measurement and render the test uninterpretable.
The sensitivity of edrophonium testing has been estimated to be about 86% for ocular MG and 95% for generalized MG [ 15 ]. False positive edrophonium testing may occur in other neurological conditions including lower motor neuron disorders and brainstem tumors [ 16 – 19 ].
During testing, up to 10 mg of intravenous edrophonium chloride may be administered. Because of the potential for serious muscarinic side effects including bronchospasm and bradycardia, atropine should be readily available. Typical muscarinic side effects include increased sweating, lacrimation, salivation, nausea, and diarrhea. An incremental dosing schedule should be utilized with one minute observation periods following each dose of edrophonium. If muscle strength improves clearly within one minute following any dose increment, the test is considered to be positive and the procedure is concluded. This strategy reduces the risk of giving excessive edrophonium and producing untoward muscarinic side effects. An initial 2 mg dose and subsequent doses of 2 mg, 3 mg, and 3 mg are given if needed.
Electrophysiologic testing
Rns studies.
With low rates of motor nerve stimulation (2–5 Hz), RNS depletes the immediate stores of acetylcholine at the neuromuscular junction. This reduces the safety factor and probability of successful neuromuscular transmission. In neuromuscular junction disorders, the safety factor is reduced, and further reduction by RNS causes some endplate potentials to fail to reach depolarization threshold. This results in a failure to elicit muscle fiber action potentials. With a reduced number of individual muscle fiber action potentials, the compound muscle action potential (CMAP) becomes reduced in both amplitude and area with a resulting decremental response.
In MG, RNS study findings are abnormal when the amplitude of the fourth CMAP is reduced more than 10% from the baseline value. This may not be present in stimulus trains recorded following rest, but it may only develop in trains collected subsequent to an exercise period as a consequence of postactivation exhaustion. The sensitivity of RNS is increased when recordings are made from clinically weak muscles. Careful attention to proper technique is important to avoid erroneous results. There must be adequate immobilization of the stimulating and recording electrodes, delivery of supramaximal stimuli, muscle warming to 35°C, and withholding of acetylcholinesterase inhibitors for at least 12 hours prior to testing. In general, proximal muscles including facial muscles, trapezius, deltoid, and biceps brachii are more likely to exhibit abnormal findings. In MG, when RNS studies are performed in a hand and in a shoulder muscle, the overall sensitivity is approximately 60%. RNS studies are relatively more sensitive in generalized MG and relatively less sensitive in ocular MG [ 20 ].
SFEMG is the most sensitive diagnostic test for detecting abnormal neuromuscular transmission. In SFEMG, individual muscle fiber action potentials generated by the same motor neuron are recorded by a specialized concentric needle with a 25 μ m diameter recording surface and a 500 Hz high-frequency filter. In most normal muscles, this arrangement facilitates recordings from two individual muscle fiber action potentials. The variability in time interval between the firing of one muscle fiber potential with relation to the other is termed the neuromuscular jitter [ 21 ].
SFEMG should be performed in a clinically weak muscle whenever possible. In many laboratories, the extensor digitorum communis (EDC) is studied initially. If the findings are normal in the EDC, a facial muscle should be studied [ 22 ]. When a facial and a limb muscle are studied, SFEMG is over 97% sensitive for detecting MG [ 23 ]. A finding of normal jitter in a clinically weak muscle virtually excludes MG as a cause of weakness in that muscle. However, SFEMG also demonstrates abnormal neuromuscular transmission related to other motor unit disorders including motor neuropathic and myopathic disorders. Normal SFEMG fiber density measurements can aid in distinguishing primary disorders of neuromuscular transmission from other motor unit disorders such as motor neuropathic or myopathic processes. In light of its reduced specificity, SFEMG must be performed and interpreted in the appropriate clinical context to avoid false positive results due to diseases other than those primarily affecting the neuromuscular junction. It is a time-consuming test that requires special expertise and equipment that are not available in all centers.
Serological testing
Achr antibodies.
The AChR binding antibody assay utilizes purified human skeletal AChRs incubated with patient serum immunoglobulin. The assay is very specific, and positive antibody studies confirm MG in a patient with appropriate symptoms and clinical findings. AChR binding antibodies are present in approximately 80% of patients with generalized MG, but in only 55% of patients with ocular MG [ 24 , 25 ]. About one-half of prepubertal children with MG are SN [ 26 ]. Though relatively less sensitive than SFEMG, AChR binding antibodies are highly specific for autoimmune MG. Rarely, false positive results in AChR binding antibody assays have been observed in patients with other autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and inflammatory neuropathy. False positive results have also been reported in motor neuron disease, patients with thymoma without MG, and relatives of patients with MG [ 27 ]. Some initially SN patients may seroconvert within the first several months of disease. Seroconversion may be identified in these patients by repeating the AChR binding antibody studies after six months of symptoms [ 28 ].
The AChR modulating antibody assay measures the degradation rate of labeled AChRs from cultured human myotubes. AChR blocking antibodies compete for the acetylcholine binding site or allosterically inhibit binding of radiolabelled α -bungarotoxin to the AChR [ 27 ]. The AChR modulating and blocking antibody assays are probably useful only when the AChR binding antibody assay is negative, since they increase diagnostic sensitivity only slightly. Approximately 4% of patients with negative AChR binding antibodies have an elevated AChR modulating antibody assay, and approximately 1% of patients with negative AChR binding antibodies demonstrate increased AChR blocking antibodies [ 29 ].
Anti-striated muscle antibodies
Anti-striated muscle or anti-striational antibodies react with contractile elements of skeletal muscle. They are found in 30% of patients with adult-onset MG, and they appear to be more common in patients with later disease onset [ 30 ]. These antibodies may be useful as a serological marker for thymoma in younger patients. Anti-striated muscle antibodies have been demonstrated in 80% of patients with thymoma in the absence of MG. Following thymoma resection, a rise in anti-striated muscle antibody titer may suggest recurrent tumor [ 30 ]. In one series, thymomas were demonstrated in 60% of patients with anti-striated muscle antibodies and MG beginning before age 50, and in less than 2% of patients without these antibodies [ 31 ].
MuSK antibodies
Antibodies to MuSK have been demonstrated recently in about one third of patients with generalized SN MG. Patients with MuSK MG are predominantly female and may exhibit prominent bulbar, neck, shoulder girdle, and respiratory weakness [ 1 , 2 , 4 ]. MuSK appears to facilitate clustering of AChR at the end plate region in the developing neuromuscular junction, though the role of MuSK antibodies in producing disease at mature neuromuscular junctions has not yet been defined.
Other antibodies
Antibodies against the intracellular skeletal muscle protein titin may be present in patients with thymoma, but they are also present in about half of patients with late-onset MG without thymoma [ 32 , 33 ]. Ryanodine antibodies are also associated with late-onset MG. Patients with ryanodine antibodies may exhibit severe, treatment-resistant MG associated with malignant thymomas [ 34 ]. Although the role for these antibodies in the diagnosis of MG has yet to be determined, they may have prognostic value and expedite chest imaging studies for detection of thymoma.
Other testing
Chest computerized tomography (CT) should be performed in patients with MG to exclude the presence of thymoma. Chest CT is more sensitive than plain chest radiographs for delineating anterior mediastinal masses, and chest MRI does not improve diagnostic sensitivity. Iodinated contrast agents have rarely precipitated significant worsening of myasthenic weakness [ 35 , 36 ]. Though this is an uncommon phenomenon [ 37 ], we do not routinely use iodinated contrast agents during chest CT studies performed to assess for thymoma. Since MG often co-exists with other autoimmune disorders, particularly autoimmune thyroid disease, patients should undergo thyroid function testing along with testing for other autoimmune disorders when clinically appropriate.
Pharmacologic testing with intravenous edrophonium is sensitive when performed in patients with significant ptosis or external ophthalmoparesis. RNS studies may demonstrate impaired neuromuscular transmission, especially when performed recording from clinically weak muscles, though they are relatively insensitive in ocular and in mild generalized MG. SFEMG is the most sensitive laboratory test for MG, although abnormal SFEMG findings may be seen in motor neuropathic and in myopathic disorders. Normal SFEMG findings in a clinically weak muscle exclude a diagnosis of MG. In the clinical context of fluctuating and fatigable weakness, AChR or MuSK antibodies confirm the diagnosis of MG, though nearly half of patients with ocular MG are SN.
Differential diagnosis
Differential diagnosis includes other disorders of the neuromuscular junction including Lambert Eaton syndrome, botulism, congenital myasthenic syndromes, and tick paralysis. In addition, acute inflammatory demyelinating polyradiculoneuropathy (AIDP) and variants of AIDP affecting cranial muscles such as the Miller-Fisher and cervical-brachial-pharyngeal variants may simulate MG, though the weakness does not have the same variability. Mitochondrial neuromuscular disorders, particularly those featuring external ophthalmoplegia and ptosis, may simulate MG. However, the onset of symptoms is gradual, and the weakness does not fluctuate greatly. Motor neuron disease involving oropharyngeal weakness may appear similar to MG, though the presence of corticobulbar features and increased fiber density measurements on SFEMG can assist in distinguishing these entities. Finally, brainstem ischemia may simulate the fluctuating character of MG, though unlike MG, consciousness, balance and coordination, and sensation are often impaired.
Treatment must be individualized to each patient with MG. The overall goal is to reestablish or to approximate normal clinical neuromuscular function while minimizing adverse effects. Few treatments have been subjected to rigorous, prospective, placebo-controlled study in MG. Factors to be considered in selecting treatment include the distribution, duration, and severity of myasthenic weakness and functional impairment, the risk for treatment complications related to medical comorbidities, age, and gender, and the ability of the patient to obtain medication and comply with drug dosing schedules and toxicity monitoring. In general, the increased risks related to long-term immune modulation become more acceptable in more severe MG to offset increased morbidity and mortality related to uncontrolled disease. A detailed review of treatment issues in autoimmune MG may be found elsewhere [ 38 ].
Acetylcholinesterase inhibitors
Acetylcholinesterase inhibitors slow the hydrolysis of acetylcholine at the neuromuscular junction and provide temporary improvement in strength in many patients with MG. Although acetylcholinesterase inhibitors were among the earliest and remain one of the most widely prescribed treatments for MG, there are no controlled clinical trials of these agents in MG. Acetylcholinesterase inhibitors are a symptomatic therapy for MG and do not retard the underlying autoimmune attack on the neuromuscular junction. The roles for acetylcholinesterase inhibitors in MG include treatment of ocular and mild generalized disease, treatment in patients who cannot receive immune suppression, and adjunctive treatment for patients receiving immunotherapy with residual or refractory myasthenic weakness.
Effective dosing of acetylcholinesterase inhibitors reduces myasthenic weakness, minimizes muscarinic medication side effects, and must be individualized to each patient's distribution of weakness and diurnal symptom fluctuation. For example, patients with prominent dysphagia may benefit by taking pyridostigmine 30 minutes before meals and those with more symptoms in the afternoon and evening may shift their dosing to later in the day to better target symptoms. Pyridostigmine bromide is generally better tolerated than neostigmine bromide due to fewer gastrointestinal side effects. A long-acting form of pyridostigmine bromide is available, though it is absorbed irregularly and tends to be overdosed. Initial dosing of pyridostigmine bromide is usually 30 mg three times a day and may be advanced to 90 mg three to four times a day. Improvement in strength begins about 20 to 30 minutes after ingestion. Peak improvement is usually observed at about 45 minutes after ingestion, and benefits may last four hours or more.
The most salient side effects of acetylcholinesterase inhibitors relate to increased muscarinic activity and include nausea, vomiting, abdominal cramping, diarrhea, diaphoresis, and increased lacrimation, salivation, and bronchial secretions. These dose dependent and self-limited side effects may be treated with glycopyrrolate, and the gastrointestinal side effects may be treated with diphenoxylate hydrochloride with atropine or loperamide hydrochloride.
Cholinergic crisis may develop with excessive dosing of acetylcholinesterase inhibitors in patients with more severe MG. In cholinergic crises, depolarization blockade at diseased neuromuscular junctions results in increased weakness, and increased muscarinic activity generates copious oropharyngeal and bronchial secretions that may obstruct the airway or be aspirated. Signs of cholinergic crisis include weakness indistinguishable from myasthenic weakness, muscle fasciculations, and symptoms of increased muscarinic activity including bradycardia.
Corticosteroids
Corticosteroids are the most widely used immune modulating agents for MG. Although the mechanism of action in MG is unknown, corticosteroids have numerous effects on the immune system including reduction of cytokine production [ 39 ]. Corticosteroids are often used as the initial immunotherapy in patients with ocular and generalized MG, particularly in patients with unsatisfactory responses to acetylcholinesterase inhibitors. These agents may produce rapid improvement in MG, though they are often associated with the liability of significant dose-dependent side effects and occasionally elicit transient and potentially serious myasthenic exacerbations within the first two weeks of treatment. Prednisone treatment produced significant improvement in strength within two to three weeks in retrospective studies of MG [ 40 , 41 ], and a Cochrane review cites significant short term benefit in MG with corticosteroids [ 42 ]. Marked improvement or remission was achieved in 80% MG patients in one large series with a 3.1 month mean time to marked improvement and a median time to maximum benefit between five and six months [ 40 ].
The most reliable clinical responses to corticosteroids occur with a high-dose daily regimen that is gradually tapered based on clinical improvement in strength. The initial prednisone dose is typically 60–80 mg/day or 1.5–2.0 mg/kg/day. This initial dose is maintained for two to four weeks, and strength is reassessed. If strength is definitely improved, dosing is transitioned to an alternate day schedule of 100–120 mg/alternate day to minimize adrenal suppression. Occasional patients are unable to tolerate an alternating-day regimen due to mood instability, variation in MG, or difficult glycemic control in occasional diabetic patients.
Myasthenic relapses may be delayed for three weeks after reductions in corticosteroid dosing, and rapid tapering may precipitate myasthenic exacerbations or crises. Therefore, corticosteroid tapering must be slow, judicious, and preceded by clinical reassessment of strength. Patients are reassessed at four to eight week intervals, and if they have maintained or improved strength and no recurrent symptoms, the prednisone dose is tapered by 10 mg/alternate day to 30 mg/alternate day, and then by 5 mg/alternate day to 20 mg/alternate day. Subsequent tapering by 2.5 mg/alternate day should be performed over longer intervals of at least 12 weeks, since many patients will begin to experience recurrent symptoms in this dosing range unless they are being treated with another immune modulating agent.
The adverse effects of corticosteroids are numerous, well known, and largely dose-dependent. These include: hypertension, fluid retention, weight gain, potassium loss, hyperlipidemia, diabetes mellitus, osteoporosis, gastric ulceration, cataracts, glaucoma, moon facies, obesity, acne, skin friability, juvenile growth suppression, and mood/personality changes. Individuals at particular risk for side effects include those who are diabetic or glucose intolerant, obese, hypertensive, osteoporotic or post-menopausal, and those with affective or thought disorders. An alternative immune modulator may be considered in such patients.
A high protein, low fat, low carbohydrate, low sodium diet is recommended to prevent untoward weight gain, hyperlipidemia, and fluid retention. Serum electrolytes, glucose, blood pressure, and weight are monitored periodically during treatment. To minimize osteopenia, calcium carbonate 1500 mg/day and vitamin D 600 IU/day are recommended. In post-menopausal women, a baseline bone density evaluation is performed and repeated every six months during treatment. Before initiating treatment with corticosteroids or any long-term immunotherapy, PPD testing should be considered as a screen for tuberculosis.
Corticosteroid-related MG exacerbations may produce transient but potentially serious increases in myasthenic weakness in up to 15% of patients beginning treatment with corticosteroids [ 40 ]. The increased weakness develops within 7–10 days after treatment begins and may last for up to one week before strength improves [ 40 , 43 ]. Patients at greatest risk for morbidity related to this phenomenon are those with more severe bulbar or generalized weakness and/or compromised respiratory function. When beginning corticosteroid treatment in such patients, strength and respiratory function should be closely monitored. Plasma exchange (PEX) may be performed prior to starting corticosteroids to circumvent or minimize corticosteroid-related MG exacerbations. In such cases, corticosteroids are initiated after a clear improvement in strength attributable to PEX is documented.
An alternative dosing strategy of starting corticosteroids at a low initial dose with gradual dose increases has been advocated to reduce the risk for corticosteroid-related MG exacerbations [ 44 ]. However, this strategy does not eliminate the risk for exacerbation [ 45 ], and onset of strength improvement is less predictable and may be significantly delayed.
Azathioprine
Azathioprine is hepatically converted to 6-mercaptopurine, an active anti-metabolite that blocks nucleotide synthesis and T-lymphocyte proliferation. Azathioprine is an effective agent for long-term immune modulation in MG as a steroid sparing drug or as initial immunotherapy. Compared to corticosteroids, azathioprine has a favorable side effect profile for long-term use. However, the typically long delay of four to eight months from beginning treatment with azathioprine to improved strength in MG is a significant liability to its usefulness, particularly in MG patients with progressive disease or functionally limiting symptoms.
In a prospective, randomized, double-blind study comparing prednisolone with prednisolone plus azathioprine, the prednisolone plus azathioprine treatment group exhibited longer remissions, fewer treatment failures, fewer side effects, and reduced maintenance doses of prednisolone [ 46 ]. The initial dose is 50 mg/day is increased by 50 mg/day every week to a target therapeutic dose of 2–3 mg/kg/day.
Side effects include dose dependent myelosuppression with macrocytic anemia, leukopenia, and thrombocytopenia, toxic hepatitis, and alopecia. Hypersensitivity pancreatitis represents a rare, but serious idiosyncratic reaction, and patients with sustained abdominal pain taking azathioprine should be screened with serum amylase and lipase assays. With long-term use, there is a small increased risk for lymphoma [ 47 ].
Azathioprine is potentially teratogenic, and women of child bearing potential using azathioprine should practice effective contraception. An idiosyncratic allergic reaction with rash, fever, nausea, vomiting, and abdominal pain occurs in 10–15% patients within the first three weeks of treatment [ 48 , 49 ]. The reaction resolves within one day of stopping azathioprine, and will recur if the patient is rechallenged with the drug.
Monitoring for myelosuppression is recommended with weekly blood count and liver transaminase measurements weekly for the first month of treatment, then monthly for the first year, then every three months thereafter unless the dosage is increased. Erythrocyte macrocytosis is expected and acceptable within the therapeutic dosage range. If white blood cell count (WBC) falls below 3500/mm 3 , the dosage should be reduced, and if WBC falls below 3000/mm 3 , azathioprine should be discontinued.
Cyclosporine
Cyclosporine exerts an immunomodulatory effect by blocking interleukin-2 production and T lymphocyte proliferation. Although effective, the use of cyclosporine in MG has been limited by its nephrotoxicity and numerous drug interactions. In light of this, cyclosporine is used in MG as a steroid-sparing agent or for refractory generalized disease. After therapeutic levels of cyclosporine are achieved and maintained, improvement in strength is usually observed within two months.
In a population of steroid-dependent patients with MG, a randomized, double blind, placebo-controlled study of cyclosporine demonstrated significantly improved strength in the cyclosporine treatment group [ 50 ]. In a long-term retrospective study, 96% MG patients improved with cyclosporine treatment, and steroids could be tapered or discontinued in 95% patients [ 51 ]. The typical cyclosporine dose is 2.5 mg/kg every 12 hours to achieve a target serum trough level of 100–150 microgram/L.
Side effects of cyclosporine include hypertension, nephropathy, tremor, hirsutism, gingival hypertrophy, headaches, and nausea. Accordingly, relative contraindications to cyclosporine include poorly controlled hypertension, renal insufficiency or failure, malignancy, and inability to comply with blood monitoring or medication precautions.
Periodic monitoring is necessary to achieve therapeutic trough cyclosporine levels and to prevent nephrotoxicity. Assessments of blood pressure, serum creatinine and the trough serum cyclosporine level should be performed frequently until a stable, therapeutic dose of cyclosporine is achieved and after new medications are begun that have the potential to interact with cyclosporine.
Cyclosporine has problematic interactions with numerous drugs that may result in nephrotoxicity, significant increases in serum drug levels, and/or significant increases or decreases in serum cyclosporine levels [ 52 ]. The most common cyclosporine interactions and potential complications include non-steroidal anti-inflammatory agents with impaired renal function, angiotensin converting enzyme inhibitors eliciting hyperkalemia, and HMG CoA reductase inhibitors precipitating cholesterol-lowering agent myopathy.
Mycophenolate mofetil
Mycophenolate mofetil (MMF) is a relatively novel immune modulator that selectively inhibits T and B lymphocyte proliferation by blocking purine synthesis exclusively in lymphocytes. In human kidney transplant trials, MMF exhibited minimal toxicity [ 53 ]. Given a paucity of side effects, MMF is used in MG both as a steroid-sparing agent and as initial immunotherapy in patients at risk for corticosteroid complications. Improved strength is observed within about two months after reaching a therapeutic dose of MMF.
Significantly improved strength in MG patients taking MMF has been demonstrated in a retrospective case series [ 54 ], in an open label pilot study in steroid dependent or refractory MG [ 55 ], and in a double-blind placebo control pilot trial in MG [ 56 ].
However, in a recently concluded multi-center, randomized, controlled trial of low dose prednisone versus low dose prednisone with MMF, there was no clinically significant benefit in MG patients treated with MMF combined with low dose prednisone beyond that observed in MG patients treated with low dose prednisone only during the initial three month trial period. Analysis of the open label phase of this study is currently pending [ 57 ].
The standard MMF dosage in MG is 1000–1500 mg twice a day. Higher doses are associated with myelosuppression, and periodic blood counts are performed during treatment as surveillance against leukopenia or anemia. There is a small increased risk for lymphoproliferative disorders in transplant patients, and a case of central nervous system (CNS) lymphoma has been documented in a patient with MG after three years of MMF treatment [ 58 ]. Side effects are relatively mild. In one series, diarrhea, abdominal pain, and nausea were reported in 27% patients, though only 6% patients discontinued MMF due to these side effects [ 59 ].
Plasma exchange
PEX is used in MG to achieve rapid, temporary improvement in strength. During PEX, plasma containing AChR antibodies is separated from whole blood and replaced by albumin or fresh frozen plasma. The procedure requires catheterizing large caliber veins. Although many patients have large antecubital veins that may be accessed serially for PEX procedures, some patients require placement of large bore dual lumen central venous catheters. PEX is generally reserved for short-term treatment to achieve rapid strength improvement in myasthenic exacerbations or crises, to prepare patients for thymectomy or another surgical procedure, to prevent steroid-related MG exacerbations in susceptible patients beginning corticosteroid treatment, and for rare patients refractory to all other treatments. An National Institutes of Health (NIH) consensus statement supports use of PEX in these instances [ 60 ]. Although there have been no controlled clinical trials of PEX in MG, several series demonstrate significantly improved strength in most patients with severe MG undergoing PEX [ 61 – 63 ]. PEX trials in MG are summarized in a Cochrane review [ 64 ].
Typically, a PEX series consists of five to six exchanges of two to three liters on alternate days. Most PEX complications are related to issues of vascular access, particularly to complications of large bore central venous catheters. Excessive fluid volume shifts during the procedure may result in hypotension or fluid overload and congestive heart failure. Sepsis and hypotension are contraindications to PEX.
Improved strength is generally observed after the second or third exchange procedure in most MG patients. Unless another form of treatment is employed, the improved strength is temporary and lasts only a few weeks at best.
Intravenous immunoglobulin
IVIg has been utilized in a number of autoimmune neuromuscular disorders including acute and chronic inflammatory polyneuropathy. It is thought to act by down regulation of autoantibodies and/or induction of anti-idiotypic antibodies. In MG, IVIg may provide short-term improvement in strength for MG exacerbations and crises, for surgical preparation in patients who are poor PEX candidates because of vascular access issues, and in patients with septicemia.
A number of studies demonstrate efficacy for IVIg in MG. A small randomized, controlled trial of IVIg 1.2 and 2.0 gm/kg compared with PEX in MG patients with exacerbation or crisis showed comparable efficacy between the PEX and IVIg treatment groups [ 65 ]. Another retrospective multicenter study demonstrated that PEX was better than IVIg in ability to successfully extubate patients in crisis at two weeks and in functional outcome at one month [ 66 ]. However, the PEX groups in both studies sustained more cardiovascular and infectious complications. Although the magnitude of responses appears to be comparable between PEX and IVIg, treatment failures have been reported to IVIg with subsequent response to PEX [ 67 ]. IVIg trials in MG are summarized in a Cochrane review [ 68 ]. The time to improved strength following IVIg infusions is somewhat variable, as demonstrated by a trial of IVIg given to prepare MG patients for thymectomy in which maximal response was delayed by up to 19 days [ 69 ]. IVIg is generally administered as 10% solution, and the standard dosage is 2 gm/kg over two to five days. However, one randomized trial found no added benefit for doses of 2 gm/kg over 1 gm/kg for MG exacerbations [ 70 ]. Pretreatment with acetaminophen and diphenhydramine may reduce the frequency and severity of idiosyncratic reactions.
Side effects include volume overload, particularly for patients with cardiomyopathy or valvular heart disease, solute-induced renal failure, especially in patients with preexisting renal insufficiency or diabetic nephropathy [ 71 ], and idiosyncratic reactions such as fever, chills, nausea, vomiting, vascular headaches, and aseptic meningitis. High infusion rates may be associated with thrombosis and stroke [ 72 ]. Serum immunoglobulin quantitation to screen for IgA deficiency is recommended, since IVIg preparations contain traces of IgA.
Thymectomy has been widely performed in an effort to achieve medication-free remission in MG following Blalock's early observations of remissions following thymectomy in non-thymomatous MG [ 73 , 74 ]. To date, there have been no prospective, randomized studies completed to assess the technique or effectiveness of thymectomy in non-thymomatous MG. Fortunately, a large, international multicenter trial is currently enrolling subjects to assess the benefit of thymectomy in non-thymomatous MG [ 75 , 76 ]. An evidence-based review was recently performed to address the role of thymectomy in the management of MG, and outcomes of thymectomy in controlled, nonrandomized studies were systematically reviewed [ 77 ]. Although patients undergoing thymectomy in non-thymomatous MG were more likely to achieve medication-free remission, become asymptomatic, or exhibit clinical improvement, the association between thymectomy and improved outcomes could attribute either to thymectomy or to differences in the study populations. Therefore, in non-thymomatous MG, thymectomy should be considered as an option to increase the probability of remission or improvement [ 77 ]. The response to thymectomy is not immediate and may be delayed for several years [ 78 – 80 ].
Controversies related to thymectomy in non-thymomatous MG include the ideal timing of the procedure with respect to MG onset, course, and patient age, the optimal surgical technique, whether patients with exclusively ocular MG should undergo thymectomy, and whether patients with SN or MuSK MG benefit from thymectomy.
Because of increased surgical risk and reduced life span, thymectomy is rarely performed in patients at greater than age 60 years for non-thymomatous MG. There is evidence to suggest that the best clinical responses occur if thymectomy is performed early in the course of MG [ 81 ], though this may attribute to a non-linear remission rate [ 77 ], as remission is more likely to occur shortly after diagnosis rather than with more chronic disease [ 82 ]. The role for thymectomy in non-thymomatous ocular MG is also uncertain [ 83 , 84 ]. In MuSK MG, no thymomas have been reported to date, and findings in recent clinical series raise doubt about benefits of thymectomy in MuSK MG patients [ 1 , 2 , 4 ].
Thymectomy may be performed via several approaches. Though more invasive than other approaches, a combined transsternal-transcervical technique is considered to be optimal as it provides the widest exposure for complete removal of thymic tissue that may be widely distributed in mediastinum and neck [ 85 ]. Surgical techniques for thymectomy have been reviewed by Jaretzki and colleagues [ 86 ]. Preoperative PEX or IVIg is performed to improve strength in patients with moderate or severe generalized or bulbar MG or in patients with MG-related respiratory insufficiency [ 87 ]. In contemporary series of thymectomy for MG, mortality is less than 1%. Complications include respiratory failure due to myasthenic crisis in 6%, infection in 11%, and recurrent laryngeal or phrenic nerve injury in 2% [ 88 ].
Most patients develop initial symptoms of extraocular muscle weakness with asymmetric ptosis and diplopia. The course is frequently variable, particularly within the first year of disease. Nearly 85% of patients with initial ocular symptoms progress to develop weakness of bulbar and limb muscles within the first three years [ 10 , 89 ]. Initial presentations with oropharyngeal and limb weakness are less common. Maximum disease severity is reached within the first year in almost two-thirds of patients [ 11 ]. Myasthenic crisis, or respiratory failure due to myasthenic weakness occurs in about 20% of patients, usually within the first year of illness [ 90 , 91 ]. Myasthenic symptoms and signs may worsen in the setting of systemic illness, particularly viral upper respiratory infections, thyroid disease, pregnancy, increased body temperature, and exposure to drugs that impair neuromuscular transmission (Table 1 ) [ 23 , 92 ].
Early in the course of MG, symptoms may fluctuate and occasionally remit, although such remissions are rarely permanent [ 89 , 93 ]. Three major stages of generalized MG have been proposed [ 94 ]. An active stage characterized by relapses and remissions lasting approximately seven years is followed by an inactive stage lasting about 10 years. The inactive stage is characterized by less disease volatility, though patients may experience exacerbations related to intercurrent illnesses, pregnancy, or exposure to medications that compromise neuromuscular transmission. In the ultimate stage of "burned-out" disease, untreated weakness may become fixed in association with muscle atrophy.
Prior to the widespread use of immunomodulators, prognosis for patients with MG was grim with about 30% mortality [ 89 ]. Along with advances in mechanical ventilation and intensive care, immunotherapy has been one of the major factors contributing to improved outcome in MG, and contemporary disease-specific mortality is less than 5% [ 95 ].
Unresolved questions
There are several contemporary issues related to MG that remain to be resolved. Issues pertaining to MuSK MG include determining the pathophysiologic role of MuSK antibodies in the development of MG, whether the immunological attack on the endplate region in MuSK MG is similar to that of SP MG, and whether thymectomy benefits patients with MuSK MG. The benefit of thymectomy in non-thymomatous SP MG is poorly defined at present, and an international, multicenter trial is currently being undertaken to determine whether and to what degree thymectomy is beneficial in non-thymomatous disease [ 76 ]. Whether corticosteroid treatment begun early in the course of ocular MG can prevent generalization has yet to be demonstrated. Finally, it remains to be determined whether and under what circumstances can immune modulation be discontinued without significant risk of relapse in MG patients that have achieved remission with immunotherapy.
Evoli A, Tonali PA, Padua L, Lo Monaco M, Scuderi F, Batocchi AP, Marino M, Bartoccioni E: Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. 2003, 126: 2304-2311. 10.1093/brain/awg223.
Article PubMed Google Scholar
Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A: Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001, 7: 365-368. 10.1038/85520.
Article CAS PubMed Google Scholar
McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, Vincent A: Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004, 55: 580-584. 10.1002/ana.20061.
Sanders DB, El-Salem K, Massey JM, McConville J, Vincent A: Clinical aspects of MuSK antibody positive seronegative myasthenia gravis (SNMG). Neurology. 2003, 60: 1978-1980.
Scuderi F, Marino M, Colonna L, Mannella F, Evoli A, Provenzano C, Bartoccioni E: Anti-P110 autoantibodies identify a subtype of "seronegative" myasthenia gravis with prominent oculobulbar involvement. Lab Invest. 2002, 82: 1139-1146.
Jaretzki A, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, Sanders DB: Myasthenia gravis: Recommendations for clinical research standards. Neurology. 2000, 55: 16-23.
Phillips LH: The epidemiology of myasthenia gravis. Ann NY Acad Sci. 2003, 998: 407-412. 10.1196/annals.1254.053.
Phillips LH, Torner JC: Epidemiologic evidence for a changing natural history of myasthenia gravis. Neurology. 1996, 47: 1233-1238.
Aragones JM, Bolibar I, Bonfill X, Mummany A, Alonso F, Illa I: Myasthenia gravis. A higher than expected incidence in the elderly. Neurology. 2003, 60: 1024-1026. 10.1001/archneur.60.7.1024-a.
Grob D: Course and management of myasthenia gravis. JAMA. 1953, 153: 529-532.
Article CAS Google Scholar
Grob D, Brunner NG, Namba T: The natural course of myasthenia gravis and effects of therapeutic measures. Ann NY Acad Sci. 1981, 377: 652-669. 10.1111/j.1749-6632.1981.tb33764.x.
Andrews PI: Autoimmune myasthenia gravis in childhood. Semin Neurol. 2004, 24: 101-110. 10.1055/s-2004-829591.
Santa T, Engel AG, Lambert EH: Histomeric study of neuromuscular junction ultrastructure I. Myasthenia gravis. Neurology. 1972, 22: 71-82.
Namba T, Brunner NG, Grob D: Myasthenia gravis in patients with thymoma, with particular reference to onset after thymectomy. Medicine. 1978, 57: 411-433. 10.1097/00005792-197809000-00002.
Phillips LH, Melnick PA: Diagnosis of myasthenia gravis in the 1990s. Semin Neurol. 1990, 10: 62-69.
Dirr LY, Donofrio PD, Patton JF, Troost BT: A false-positive edrophonium test in a patient with a brainstem glioma. Neurology. 1989, 39: 865-867.
Moorthy G, Behrens MM, Drachman DB, Kirkham TH, Knox DL, Miller NR, Slamovitz TL, Zinreich SJ: Ocular pseudomyasthenia or ocular myasthenia "plus": A warning to clinicians. Neurology. 1989, 39: 1150-1154.
Mulder DW, Lambert EH, Eaton LM: Myasthenic syndrome in patients with ALS. Neurology. 1959, 9: 627-631.
Ragge NK, Hoyt WF: Midbrain myasthenia: fatigable ptosis, lid twitch sign, and ophthalmoparesis from a dorsal midbrain glioma. Neurology. 1992, 42: 917-919.
Howard JF, Sanders DB, Massey JM: The electrodiagnosis of myasthenia gravis and the Lambert-Eaton myasthenic syndrome. Neurol Clin. 1994, 12: 305-330.
PubMed Google Scholar
Stålberg EV, Trontelj JV: Single fiber electromyography: studies in healthy and diseased muscle 2nd edition. New York: Raven Press; 1994.
Google Scholar
AAEM Quality Assurance Committee, American Association of Electrodiagnostic Medicine: Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: Summary statement. Muscle Nerve. 2001, 24: 1236-1238. 10.1002/mus.1139.
Article Google Scholar
Sanders DB, Howard JF: Disorders of neuromuscular transmission. Neurology in clinical practice. 1st edition. Edited by: Bradley WG, Daroff RB, Fenichel GM, Marsden CD. Boston: Butterworth-Heinemann; 1991:1819-1842.
Lindstrom J: An assay for antibodies to human acetylcholine receptor in serum from patients with myasthenia gravis. Clin Immunol Immunopathol. 1977, 7: 36-43. 10.1016/0090-1229(77)90027-7.
Vincent A, Newsom-Davis J: Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry. 1985, 48: 1246-1252.
Article PubMed Central CAS PubMed Google Scholar
Andrews PI, Massey JM, Sanders DB: Acetylcholine receptor antibodies in juvenile myasthenia gravis. Neurology. 1993, 43: 977-982.
Lennon VA: Serological diagnosis of myasthenia gravis and Lambert-Eaton myasthenic syndrome. Handbook of Myasthenia Gravis and Myasthenic Syndromes. Edited by: Lisak RP. New York: Marcel-Dekker; 1994:149-164.
Sanders DB, Andrews I, Howard JF, Massey JM: Seronegative myasthenia gravis. Neurology. 1997, 48 (Suppl 5): S40-S45.
Howard FM, Lennon VA, Finley J, Matsumoto J, Elveback LR: Clinical correlations of antibodies that bind, block, or modulate human acetylcholine receptors in myasthenia gravis. Ann NY Acad Sci. 1987, 505: 526-538. 10.1111/j.1749-6632.1987.tb51321.x.
Cikes N, Momoi MY, Williams CL, Howard FM, Hoagland HC, Whittingham S, Lennon VA: Striational autoantibodies: Quantitative detection by enzyme immunoassay in myasthenia gravis, thymoma, and recipients of D-penicillamine or allogeneic bone marrow. Mayo Clin Proc. 1988, 63: 474-481.
Sanders DB, Massey JM: The diagnostic utility of anti-striational antibodies in myasthenia gravis [abstract]. Neurology. 2002, 58: A229-10.1159/000066266.
Somnier FE, Engel PJ: The occurrence of anti-titin antibodies and thymomas: A population survey of MG 1970–1999. Neurology. 2002, 59: 92-98.
Yamamoto AM, Gajdos P, Eymard B, Tranchant C, Warter JM, Gomez L, Bourquin C, Bach JF, Garchon HJ: Anti-titin antibodies in myasthenia gravis. Tight association with thymoma and heterogeneity of nonthymoma patients. Arch Neurol. 2001, 58: 885-890. 10.1001/archneur.58.6.885.
Mygland A, Aarli JA, Matre R, Gilhus NE: Ryanodine receptor antibodies related to severity of thymoma associated myasthenia gravis. J Neurol Neurosurg Psychiatry. 1994, 57: 843-846.
Chagnac Y, Hadanin M, Goldhammer Y: Myasthenic crisis after intravenous administration of iodinated contrast agent. Neurology. 1985, 35: 1219-1220.
Canal N, Franceschi M: Myasthenic crisis precipitated by iothalamic acid. Lancet. 1983, 1: 1288-10.1016/S0140-6736(83)92749-6.
Frank JH, Cooper GW, Black WC, Phillips LH: Iodinated contrast agents in myasthenia gravis. Neurology. 1987, 37: 1400-1402.
Juel VC, Massey JM: Autoimmune myasthenia gravis: Recommendations for treatment and immunologic modulation. Curr Treat Options Neurol. 2005, 7: 3-14. 10.1007/s11940-005-0001-7.
McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, Goldfarb RH, Kitson RP, Miller AH, Spencer RL, Weiss JM: The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Rev. 1997, 23: 79-133. 10.1016/S0165-0173(96)00012-4.
Pascuzzi RM, Coslett HB, Johns TR: Long-term corticosteroid treatment of myasthenia gravis: Report of 116 patients. Ann Neurol. 1984, 15: 291-298. 10.1002/ana.410150316.
Johns TR: Long-term corticosteroid treatment of myasthenia gravis. Ann NY Acad Sci. 1987, 505: 568-583. 10.1111/j.1749-6632.1987.tb51325.x.
Schneider-Gold C, Gajdos P, Toyka KV, Hohlfeld RR: Corticosteroids for myasthenia gravis. Cochrane Database of Systematic Reviews. 2005, 2: CD002828.
Miller RG, Milner-Brown HS, Mirka A: Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology. 1986, 36: 729-732.
Seybold ME, Drachman DB: Gradually increasing doses of prednisone in myasthenia gravis: reducing the hazards of treatment. N Engl J Med. 1974, 290: 81-84.
Sghirlanzoni A, Peluchetti D, Mantegazza R, Fiacchino F, Cornelio F: Myasthenia gravis: prolonged treatment with steroids. Neurology. 1984, 34: 170-174.
Palace J, Newsom-Davis J, Lecky B: A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology. 1998, 50: 1778-1783.
Herrllinger U, Weller M, Dichgans J, Melms A: Association of primary central nervous system lymphoma with long-term azathioprine therapy for myasthenia gravis. Ann Neurol. 2000, 47: 682-683. 10.1002/1531-8249(200005)47:5<682::AID-ANA24>3.0.CO;2-Z.
Hohlfeld R, Michels M, Heininger K, Besinger U, Toyka KV: Azathioprine toxicity during long-term immunosuppression of generalized myasthenia gravis. Neurology. 1988, 38: 258-261.
Kissel JT, Levy RJ, Mendell JR, Griggs RC: Azathioprine toxicity in neuromuscular disease. Neurology. 1986, 36: 35-39.
Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K: A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann NY Acad Sci. 1993, 681: 539-551. 10.1111/j.1749-6632.1993.tb22937.x.
Ciafaloni E, Nikhar NK, Massey JM, Sanders DB: Retrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology. 2000, 55: 448-450.
Sanders DB, Howard JF: Disorders of neuromuscular transmission. Neurology in clinical practice. 3rd edition. Edited by: Bradley WG, Daroff RB, Fenichel GM, Marsden CD. Boston: Butterworth-Heinemann; 2000, 2167-2185.
Sollinger HW, Renal Transplant Mycophenolate Mofetil Study Group: Mycophenolate mofetil for the prevention of acute rejection in primary cadaveric renal allograft recipients. U.S. Renal Transplant Mycophenolate Mofetil Study Group. Transplantation. 1995, 60: 225-232. 10.1097/00007890-199508000-00003.
Chaudhry V, Cornblath DR, Griffin JW, O'Brien R, Drachman DB: Mycophenolate mofetil: A safe and promising immunosuppressant in neuromuscular diseases. Neurology. 2001, 56: 94-96.
Ciafaloni E, Massey JM, Tucker-Lipscomb B, Sanders DB: Mycophenolate mofetil for myasthenia gravis: An open-label pilot study. Neurology. 2001, 56: 97-99.
Meriggioli MN, Rowin J, Richman JG, Leurgans S: Mycophenolate mofetil for myasthenia gravis: A double-blind, placebo-controlled pilot study. Ann NY Acad Sci. 2003, 998: 494-499. 10.1196/annals.1254.064.
Sanders D, McDermott M, Thornton C, Tawil A, Barohn R, the Muscle Study Group: A trial of mycophenolate mofetil (MMF) with prednisone as initial immunotherapy in myasthenia gravis (MG) [abstract]. Neurology. 2007, 68: A107-10.1159/000110591.
Vernino S, Salomao DR, Habermann TM, O'Neill BP: Primary CNS lymphoma complicating treatment of myasthenia gravis with mycophenolate mofetil. Neurology. 2005, 65: 639-641. 10.1212/01.wnl.0000173031.56429.04.
Meriggioli MN, Ciafaloni E, Al-Hayk KA, Rowin J, Tucker-Lipscomb B, Massey JM, Sanders DB: Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology. 2003, 61: 1438-1440.
NIH Consensus Conference: The utility of therapeutic plasmapheresis for neurological disorders. JAMA. 1986, 256: 1333-1337. 10.1001/jama.256.10.1333.
Pinching AF, Peters DK, Newsom-Davis J: Remission of myasthenia gravis following plasma exchange. Lancet. 1976, 2: 1373-1376. 10.1016/S0140-6736(76)91917-6.
Dau PC, Lindstrom JM, Cassel CK, Denys EH, Shev EE, Spitler LE: Plasmapheresis and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977, 297: 1134-1140.
Antozzi C, Gemma M, Regi B, Berta E, Confalonieri P, Peluchetti D, Mantegazza R, Baggi F, Marconi M, Fiacchino F, et al: A short plasma exchange protocol is effective in severe myasthenia gravis. J Neurol. 1991, 238: 103-107. 10.1007/BF00315690.
Gajdos P, Chevret S, Toyka K: Plasma exchange for myasthenia gravis. Cochrane Database of Systematic Reviews. 2002, 4: CD002275.
Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C: Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group. Ann Neurol. 1997, 41: 789-796. 10.1002/ana.410410615.
Qureshi AI, Choudhry MA, Akbar MS, Mohammad Y, Chua HC, Yahia AM, Ulatowski JA, Krendel DA, Leshner RT: Plasma exchange versus intravenous immunoglobulin treatment in myasthenic crisis. Neurology. 1999, 52: 629-632.
Stricker RB, Kwiatkowska BJ, Habis JA, Kiprov DD: Myasthenic crisis. Response to plasmapheresis following failure of intravenous gamma-globulin. Arch Neurol. 1993, 50: 837-840.
Gajdos P, Chevret S, Toyka K: Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. 2006, 19 (2): CD002277.
Huang C-S, Hsu H-S, Kao K-P, Huang M-H, Huang B-S: Intravenous immunoglobulin in the preparation of thymectomy for myasthenia gravis. Acta Neurol Scand. 2003, 108: 136-138. 10.1034/j.1600-0404.2003.00131.x.
Gajdos P, Tranchant C, Clair B, Bolgert F, Eymard B, Stojkovic T, Attarian S, Chevret S, Myasthenia Gravis Clinical Study Group: Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin 1g/kg versus 2g/kg: A randomized double blind clinical trial. Ann Neurol. 2005, 62: 1689-1693. 10.1001/archneur.62.11.1689.
Tan E, Hajinazarian M, Bay W, Neff J, Mendel JR: Acute renal failure resulting from intravenous immunoglobulin therapy. Arch Neurol. 1993, 50: 137-139.
Caress JB, Cartwright MS, Donofrio PD, Peacock JE: The clinical features of 16 cases of stroke associated with administration of IVIg. Neurology. 2003, 60: 1822-1824.
Blalock A, Harvey AM, Ford FR, Lilienthal JL: The treatment of myasthenia gravis by removal of the thymus gland. J American Medical Association. 1941, 117: 1529-1533.
Blalock A: Thymectomy in the treatment of myasthenia gravis. Report of twenty cases. J Thorac Surg. 1944, 13: 316-339.
Wolfe GI, Kaminski HJ, Jaretzki A, Swan A, Newsom-Davis J: Development of a thymectomy trial in nonthymomatous myasthenia gravis patients receiving immunosuppressive therapy. Ann NY Acad Sci. 2003, 998: 473-480. 10.1196/annals.1254.061.
A Multi-Center, Single-Blind, Randomized Study Comparing Thymectomy to No Thymectomy in Non-Thymomatous Myasthenia Gravis (MG) Patients Receiving Prednisone (MGTX). Accessed 15 March 2007, [ http://www.soph.uab.edu/mgtx/ ]
Gronseth GS, Barohn RJ: Practice parameter: Thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000, 55: 7-15.
Perlo VP, Arnason B, Poskanzer D, Castleman B, Schwab RS, Osserman KE, Papatestis A, Alpert L, Kark A: The role of thymectomy in the treatment of myasthenia gravis. Ann NY Acad Sci. 1971, 183: 308-315. 10.1111/j.1749-6632.1971.tb30761.x.
Rodriguez M, Gomez MR, Howard FM, Taylor WF: Myasthenia gravis in children: Long-term follow-up. Ann Neurol. 1983, 13: 504-510. 10.1002/ana.410130506.
Mulder DG, Graves M, Hermann C: Thymectomy for myasthenia gravis: Recent observation and comparisons with past experience. Ann Thorac Surg. 1989, 48: 551-555.
Monden Y, Nakahara K, Kagotani K, Fujii Y, Nanjo S, Masaoka A, Kawashima Y: Effects of preoperative duration of symptoms on patients with myasthenia gravis. Ann Thorac Surg. 1984, 38: 287-291.
Beghi E, Antozzi C, Batocchi AP, Cornelio F, Cosi V, Evoli A, Lombardi M, Mantegazza R, Monticelli ML, Piccolo G, et al: Prognosis of myasthenia gravis: A multicenter follow-up study of 844 patients. J Neurol Sci. 1991, 106: 213-220. 10.1016/0022-510X(91)90260-E.
Lanska DJ: Indications for thymectomy in myasthenia gravis. Neurology. 1990, 40: 1828-1829.
Schumm F, Wietholter H, Fateh-Moghadam A, Dichgans J: Thymectomy in myasthenia with pure ocular symptoms. J Neurol Neurosurg Psychiatry. 1985, 48: 332-337.
Jaretzki A: Thymectomy for myasthenia gravis: Analysis of the controversies regarding technique and results. Neurology. 1997, 48 (Suppl 5): S52-S63.
Jaretzki A, Steinglass KM, Sonett JR: Thymectomy in the management of myasthenia gravis. Semin Neurol. 2004, 24: 49-62. 10.1055/s-2004-829596.
Jaretzki A, Aarli JA, Kaminski HJ, Phillips LH, Sanders DB: Preoperative preparation of patients with myasthenia gravis forestalls postoperative respiratory complications after thymectomy. Ann Thorac Surg. 2003, 75: 1068-10.1016/S0003-4975(02)03811-0.
Bulkley GB, Bass KN, Stephenson GR, Diener-West M, George S, Reilly PA, Baker RR, Drachman DB: Extended cervicomediastinal thymectomy in the integrated management of myasthenia gravis. Ann Surg. 1997, 226: 324-334. 10.1097/00000658-199709000-00012.
Oosterhuis HJ: The natural course of myasthenia gravis: A long-term followup study. J Neurol Neurosurg Psychiatry. 1989, 52: 1121-1127.
Sellman MS, Mayer RF: Treatment of myasthenic crisis in late life. South Med J. 1985, 78: 1208-1210.
Thomas CE, Mayer SA, Gungor Y, Swarup R, Webster EA, Chang I, Brannagan TH, Fink ME, Rowland LP: Myasthenic crisis: Clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997, 48: 1253-1260.
Pascuzzzi RM: Medications and myasthenia gravis: A reference for health care professionals. Accessed 15 March 2007, [ http://myasthenia.org/docs/MGFA_MedicationsandMG.pdf ]
Osserman KE: Myasthenia gravis. New York: Grune & Stratton; 1958:78-80.
Simpson JA, Thomaides T: Treatment of myasthenia gravis: An audit. Q J Med. 1987, 64: 693-704.
CAS PubMed Google Scholar
Grob D: Natural history of myasthenia gravis. Myasthenia gravis and myasthenic disorders. Edited by: Engel AG. 1999, New York: Oxford University Press, 131-154.
Download references
Author information
Authors and affiliations.
Division of Neurology, Duke University Medical Center, Box 3403, Durham, North Carolina, 27710, USA
Vern C Juel & Janice M Massey
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Vern C Juel .
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License ( http://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reprints and permissions
About this article
Cite this article.
Juel, V.C., Massey, J.M. Myasthenia gravis. Orphanet J Rare Dis 2 , 44 (2007). https://doi.org/10.1186/1750-1172-2-44
Download citation
Received : 23 April 2007
Accepted : 06 November 2007
Published : 06 November 2007
DOI : https://doi.org/10.1186/1750-1172-2-44
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Acetylcholinesterase Inhibitor
- Edrophonium
- Congenital Myasthenic Syndrome
- Pyridostigmine Bromide
Orphanet Journal of Rare Diseases
ISSN: 1750-1172
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]
- Open access
- Published: 20 December 2021
Global prevalence of myasthenia gravis and the effectiveness of common drugs in its treatment: a systematic review and meta-analysis
- Nader Salari 1 ,
- Behnaz Fatahi 2 ,
- Yalda Bartina 3 ,
- Mohsen Kazeminia 2 ,
- Reza Fatahian 4 ,
- Payam Mohammadi 5 ,
- Shamarina Shohaimi 6 &
- Masoud Mohammadi ORCID: orcid.org/0000-0002-5722-8300 7
Journal of Translational Medicine volume 19 , Article number: 516 ( 2021 ) Cite this article
13k Accesses
21 Citations
319 Altmetric
Metrics details
Myasthenia gravis is a neuromuscular autoimmune disorder characterized by weakness and disability in the voluntary muscles. There have been several preliminary studies on the epidemiology of myasthenia gravis in different parts of the world and the effectiveness of common drugs in its treatment, but there has been no comprehensive study of the efficacy of common drugs in the treatment of myasthenia gravis. Therefore, this study aimed to determine the epidemiology of myasthenia gravis globally and the effectiveness of common drugs in its treatment using systematic review and meta-analysis.
Research studies were extracted from IranDoc, MagIran, IranMedex, SID, ScienceDirect, Web of Sciences (WoS), ProQuest, Medline (PubMed), Scopus and Google Scholar based on Cochran's seven-step guidelines using existing keywords extracted in MeSH browser. The I 2 test was used to calculate the heterogeneity of studies, and Begg and Mazumdar rank correlation tests were used to assess publication bias. Data were analyzed using Comprehensive Meta-Analysis software (Version 2).
In the search for descriptive studies based on the research question, 7374 articles were found. After deleting articles unrelated to the research question, finally, 63 articles with a sample size of 1,206,961,907 people were included in the meta-analysis. The prevalence of MG worldwide was estimated to be 12.4 people (95% CI 10.6–14.5) per 100,000 population. For analytical studies on the effectiveness of common myasthenia gravis drugs, 4672 articles were found initially, and after removing articles unrelated to the research question, finally, 20 articles with a sample size of 643 people in the drug group and 619 people in the placebo group were included in the study. As a result of the combination of studies, the difference between the mean QMGS score index after taking Mycophenolate and Immunoglobulin or plasma exchange drugs in the group of patients showed a significant decrease of 1.4 ± 0.77 and 0.62 ± 0.28, respectively (P < 0.01).
The results of systematic review of drug evaluation in patients with myasthenia gravis showed that Mycophenolate and Immunoglobulin or plasma exchange drugs have positive effects in the treatment of MG. It also represents the positive effect of immunoglobulin or plasma exchange on reducing SFEMG index and QMGS index and the positive effect of Mycophenolate in reducing MG-ADL index, SFEMG and Anti-AChR antibodies index. In addition, based on a meta-analysis of the random-effect model, the overall prevalence of MG in the world is 12.4 people per 100,000 population, which indicates the urgent need for attention to this disease for prevention and treatment.
Myasthenia gravis (MG) is a neuromuscular disease characterized by weakness in the voluntary muscles [ 1 , 2 ]. This disease has different symptoms that vary in different patients depending on the degree of involvement of the striated muscles. The most common type of symptom in patients with myasthenia gravis is ocular symptoms, which appear as ptosis and diplopia. These symptoms usually occur at the end of the day and follow activities such as watching TV or driving is more common, and excessive fatigue has been reported due to frequent activity in patients with this disease [ 3 ].
Myasthenia gravis is an autoimmune disease that connects the nerve to the muscle (NMJ) [ 4 ], which is produced by different antibodies against synaptic membrane proteins [ 5 ]. This is usually more than 85% of cases and is caused by a type of antibody against the skeletal muscle acetylcholine receptor (AChR-Ab) [ 6 , 7 ]. However, components other than AChR, such as muscle-specific tyrosine kinase receptor (MuSK) or lipoprotein-associated protein 4 (LRP4), may also be targeted for the autoimmune attack [ 6 , 8 , 9 ].
Based on the mechanism of autoimmune disease and antibodies, invasive skeletal muscle molecules, thymus status, genetic characteristics, disease phenotype and response to treatment, myasthenia gravis is divided into early and late ocular subtypes (OMG), seronegative, thymoma, LRP4, MuSk. Diagnosis of MG subtype influences treatment decisions and disease prognosis [ 10 , 11 ]. Approximately 50% of patients with OMG develop general myasthenia gravis (GMG) over a 2-year period, which affects other muscles and manifests as weakness and ocular symptoms [ 12 ].
According to a systematic population-based study, CAR et al. [ 13 ] estimated the incidence and prevalence of MG at 54 per million and 77.7 per million, respectively. However, significant changes have been reported in various studies. The incidence of this disease has shown a range between 1.77 and 21.3 per million people and the prevalence of 15 to 179 million people [ 13 ]. A large number of epidemiological studies, mainly in Western Europe and Asia, reported significant differences in the incidence and prevalence of MG. The incidence of myasthenia gravis ranged from 1.7 to 30 per million per year [ 14 , 15 , 16 , 17 ].
The disease has two age peaks: age 40–40 years, which mainly affects women, and another 80–60 years, which occurs equally in men and women [ 4 ].
Current treatment options mainly include acetylcholinesterase inhibitors, glucocorticoids (GC), intravenous immunoglobulin (IVIg), plasma replacement (PLEX), thymectomy, and immunosuppressive agents including azathioprine prednisone, cyclosporine, cyclosporine, and cyclosporine [ 18 , 19 , 20 , 21 , 22 , 23 , 24 ]. However, the use of corticosteroids and immunosuppressants such as azathioprine manages MG. However, many patients do not tolerate or respond adequately to these drugs, and long-term treatment with GC is associated with a significant risk of side effects such as diabetes, obesity, and high blood pressure. This has led to the introduction of newer immunosuppressants such as mycophenolate mofetil (MMF) [ 12 , 25 , 26 , 27 ].
About 15% of patients with general myasthenia gravis do not respond to immunosuppressants and require intravenous immunoglobulin (IVIg) or plasma replacement (PLEX) to improve their symptoms [ 28 , 29 ]. Intensified cases and myasthenia gravis crisis also require immediate treatment due to poor swallowing or respiratory failure that threatens the lives of these patients and muscle defects that may be a major disability for their daily activities [ 30 ].
Therefore, additional immunosuppression is often treated with Plasma Freesia (PLEX) and intravenous immunoglobulin (IVIg) to relieve symptoms. Plasma freeze uses filtration to kill pathological antibodies used in patients with myasthenia gravis and severe MG [ 31 , 32 , 33 ].
Myasthenia gravis has high direct health care costs (including long-term treatment and periodic hospitalization costs) and indirect costs such as loss of income and reduced care productivity [ 34 ]. Therefore, accurate identification of patients with MG is vital for organizing health care services and implementing preventive health measures. Many early articles have been done on the prevalence of myasthenia gravis and the effect of different drugs on the treatment process, but appropriate policy to control, diagnose and treat this disease requires coherent, accurate and uniform information. Therefore, the present study was performed to estimate the prevalence of myasthenia gravis globally and determine the effectiveness of the most common drugs in the treatment of patients by systematic review and meta-analysis.
The present systematic review and meta-analysis were conducted according to the Cochrane seven-step approach, including selecting research questions, determining inclusion and exclusion criteria, identifying descriptive articles, selecting studies, qualitative evaluation of studies, data extraction, analysis and interpretation of findings [ 35 ].
Research question and determining the keywords of the descriptive section
According to the research question in the descriptive section, "How has the prevalence of MG in the world changed?" The population included: MG patients, Outcome included MG prevalence, Time or duration Included: Date of publication of the first related article until 15 November 2020 and type of study (study design) Included: cross-sectional (descriptive) studies. Keywords were extracted from the MeSH browser. Keywords related to the studied population (P): Myasthenia Gravis, MG and outcome keywords (O); Prevalence and Epidemiology.
Research question and determining the keywords of the analytical section
According to the research question of the analytical section, "What is the effectiveness of Corticosteroids, Mycophenolate and Immunoglobulin or plasma exchange drugs in the treatment of MG?" According to the PICO guidelines, the study population (Population) includes patients with MG, intervention (intervention) including Corticosteroids, Mycophenolate and Immunoglobulin or plasma exchange, analogous (Comparison) including QMGS index score, Anti-AChR antibodies, SFEMG And MG-ADL before and after the intervention, the outcome (Outcome) included: changes in QMGS, Anti-AChR antibodies, SFEMG and MG-ADL after the intervention. Keywords were extracted from the MeSH browser according to PICO instructions. Keywords related to the study population (P): Myasthenia Gravis, MG Keywords related to the intervention (I); Corticosteroids, Corticotropin, Alternate-day prednisone, Methylprednisone, Prednisolone, Mycophenolate, Immunoglobulin or plasma exchange, Intravenous immunoglobulin, IVIG and keywords related to analogy (C) and outcome (O); QMGS, QMG, Anti-AChRantibodies, Anti-AChR ab, SFEMG and MG-ADL.
Criteria for inclusion and exclusion according to the descriptive research question
Cross-sectional (descriptive) studies in chronic patients reporting the prevalence of MG in different parts of the world published in English, and the full text was available. Observational studies, cohort, case–control, analytical and interventional studies, case reports, short reports, letters to the editor and studies unrelated to the research question were excluded from the study.
Criteria for inclusion and exclusion according to the research question of the analytical section
Clinical trial studies that reported the mean and standard deviation of the effect of Corticosteroids, Mycophenolate and Immunoglobulin or plasma exchange on at least one of the indicators of QMGS, Anti-AChR antibodies, SFEMG and MG-ADL in patients with MG, in Persian and were printed in English and their full text was available and included in the study. Descriptive studies, cross-sectional studies, reviews, case reports, short reports, letters to the editor, and other studies unrelated to the research question were excluded from the study.
Articles identification
To find studies related to research questions, four Persian databases, including IranDoc, MagIran, IranMedex and SID and five international databases: ScienceDirect, Web of Science (WoS), ProQuest, Medline (PubMed), Scopus were searched. The Google Scholar scientific search engine was reviewed for final review. No time limit was set for the search to retrieve the relevant research, so all articles published by November 15, 2020, were reviewed. The search was limited to studies published in Persian and English. The search strategy in each database was determined using Advanced Search (Advanced Search) with the help of all possible keyword combinations with the help of (AND) and (OR) operators. For example, the search strategy in the PubMed database for the descriptive part of the research was determined as follows:
(((Prevalence [Title/Abstract]) OR (Epidemiology [Title/Abstract])) AND (Myasthenia Gravis [Title/Abstract])) OR (chronic patients [Title/Abstract]) OR (MG [Title/Abstract]).
Also, the search strategy in the PubMed database for the analytical part of the research was determined as follows:
((((((((((((((((Corticosteroids[Title/Abstract]) OR (Corticotropin[Title/Abstract])) OR (Alternate-day prednisone[Title/Abstract])) OR (Methylprednisone [Title/Abstract])) OR (chronic patients [Title/Abstract]) OR (Prednisolone[Title/Abstract])) OR (Mycophenolate[Title/Abstract])) OR (Immunoglobulin[Title/Abstract] OR plasma exchange[Title/Abstract])) OR (Intravenous immunoglobulin[Title/Abstract])) OR (IVIG[Title/Abstract])) AND (QMGS[Title/Abstract])) OR (QMG[Title/Abstract])) OR (Anti-AChR antibodies[Title/Abstract])) OR (Anti-AChR ab[Title/Abstract])) OR (SFEMG[Title/Abstract])) OR (MG-ADL[Title/Abstract])) AND (Myasthenia Gravis[Title/Abstract])) OR (MG[Title/Abstract]).
In order to access the latest published studies, an alert was created on several databases, including PubMed and Scopus, to check if new articles were published during the study. Also, in order to access all related studies, the sources of articles that met the inclusion criteria were manually reviewed. To avoid errors, all steps of article search, study selection, qualitative evaluation and data extraction were performed independently by two researchers (M.K. and B.F.). If there was a difference of opinion between the researchers regarding the inclusion of the article in the study, in order to avoid the risk of bias for specific studies, first a final agreement was reached through discussion and in some cases with the participation and opinion of a third party (MM).
Selection of studies based on entry and exit criteria
Based on the 4-step PRISMA process, including article identification, screening, eligibility, and inclusion in the study, three researchers reviewed this process as follows, and studies were selected based on inclusion and exclusion criteria. All articles found in each database were transferred to EndNote X8 software. After completing the search in all the databases, the articles repeated in different databases were deleted. Then, in order to avoid the risk of bias in selecting studies, the names of the authors and the titles of the journals were removed, and a checklist was prepared based on the titles and abstracts of the studies. In the next step, two authors (M.K. and B.F.) independently examined the title and abstract of the studies and eliminated studies that were not related to the research based on the inclusion and exclusion criteria of the study and in case of discrepancy, it was examined by the third researcher (M.M). Studies whose full text was not found were also excluded from the systematic review and meta-analysis process. The full text of all remaining articles was then evaluated. Studies that did not meet the inclusion criteria based on the research question were excluded.
Qualitative evaluation of descriptive studies
Qualitative evaluation of studies was performed using the STROBE checklist, a suitable tool for the qualitative assessment of descriptive studies. This checklist has 22 general items, each of which has sub-items (32 sub-items in total) and to evaluate different parts of a study, including title and abstract, study objectives, problem statement, study type, sampling method, study statistical population, the sample size is the definition of variables, tools for collecting study data, statistical analysis, findings and discussion. In order to rate the articles, if each article referred to the items considered in the checklist, it was given a score of 1, and if it was not mentioned, a score of zero was given. The minimum and maximum scores in this checklist are 0 and 32, respectively. Articles with scores of 16 and above were considered high and medium quality studies and were included in the systematic review and meta-analysis process, and articles with scores below 16 were considered low-quality studies [ 36 ].
Qualitative evaluation of analytical studies
Qualitative evaluation of studies was performed using the CONSORT checklist, a suitable tool for the qualitative assessment of interventional studies. This checklist has 25 general items, each with minor items (a total of 37 minor items). Different sections include Title and Abstract, Introduction and Background, Methods, Participants, Interventions, Objectives, Consequences, Sample Size, Randomization, How to Assign Participants, Blind Allocation, Execution, Blindness of Study, Statistical Methods, Results, Flow Participants' presence, sampling method, initial data of the number of people analyzed, consequences and estimates, auxiliary analysis, adverse reactions, explanations, interpretation, generalizability and general evidence. In this study, all general checklist items were reviewed by two authors (M.K. and B.F.). In order to rate the articles, if each article referred to the items in the checklist, it was given a score of 1, and if it was not mentioned, a score of zero was given. The minimum and maximum scores in this checklist are 0 and 37, respectively. Studies with 75% or more of the maximum achievable score (score greater than or equal to 27) with “high quality”, studies with a score between 75 and 50% (score 18–26) as “average quality” and studies with a score below 50% (less than or equal to 17) were considered “low quality” studies [ 37 ]. Based on this checklist, medium and high-quality articles were included in the study, and low-quality articles were excluded.
Data extraction
After selecting the studies to enter the systematic review and meta-analysis process, the data were extracted, and the studies were summarized. For this purpose, two electronic checklists (one for the descriptive section and one for the analytical section) were prepared. The various items in the descriptive checklist included: name of the first author, year of publication and year of the report, place of study, age, sample size and prevalence, and various items in the analytical checklist, including the name of the first author, year of publication, place of research, the sample size of the drug group and the placebo group was the type of drug, mean and standard deviation before and after the intervention.
Statistical analysis of the descriptive part
To analyze and combine the results of different studies, in each study, the prevalence of MG was considered as the probability of binomial distribution and its variance was calculated through binomial distribution. Heterogeneity of studies was assessed using the I 2 test, and the random-effects model was used in the case of the I 2 index above 50%. In this model, parametric changes between studies are also considered in the calculations, so it can be said that the results of this model in heterogeneous conditions can be more generalized than the model with a fixed effect. Publication bias assessment was performed using Funnel Plot and Begg and Mazumdar rank correlation test. Data were analyzed using Comprehensive Meta-Analysis (Version 2) software, and the significance level of the test was P < 0.05.
Statistical analysis of the analytical section
In this study, the standardized mean difference was calculated. The main outcome of this study was the mean score of the studied indicators before and after the intervention in patients with MG. As a result, the mean and standard deviation of the studied indices before and after the intervention were extracted. I 2 index was used to evaluate heterogeneity. Funnel Plot and Begg and Mazumdar rank correlation tests were used to assess the publication bias. The significance level of the test was considered 0.1. Data were analyzed using Comprehensive Meta-Analysis software (Version 2).
Descriptive part of the study
Summary of how articles enter meta-analysis: In the first stage, 7374 articles (7192 articles in international databases, 159 articles in Persian databases and 23 studies in reviewing article sources) were found, of which 5368 studies were repeated in different databases were removed. A total of 2006 studies were entered the in the screening stage and 1851 articles were deleted based on the inclusion and exclusion criteria by reviewing the title and abstract of the study. In the next stage (competency assessment), out of the remaining 155 studies from the screening stage, 92 articles were removed by reviewing the full text of the article because it was not relevant to the research. The quality evaluation of 63 articles included in this study was performed using the STROBE checklist, all of which were of medium and high quality according to the criteria of this tool. Thus, 63 articles related to the descriptive part of the study entered the process of systematic review and meta-analysis (Fig. 1 ).

Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA 2009) flow diagram Descriptive section
General characteristics of the studies
The total sample size of the studies was 1,206,961,907 people. The studies were published between 1969 and November 15, 2020. There were 8 studies in Asia, 42 in Europe, 7 in the United States, 5 in Africa, and 1 in Australia. Summary of study characteristics, including the name of the first author, year of publication and year of the report, place of study, mean age of patients, sample size and prevalence of MG, are reported in Table 1 .
Systematic meta-analysis and descriptive review
The result of the I 2 test for the prevalence of MG in the world indicates a significant heterogeneity between studies (I 2 = 99.9), so the data were analyzed by meta-analysis using a random-effects model. Due to the high heterogeneity of the studies, sensitivity analysis was performed, and each study's effect on the final result and the degree of heterogeneity was evaluated. None (P = 0.103) (Fig. 2 ). As a result of the combination of studies, the overall estimate of the global prevalence of MG 1 2 . 4 people (95% confidence interval: 10-14-5.5) per 100,000 population was based on a random-effects model. The black square is the prevalence and the length of the line segment on which the 95% confidence interval per It is a study, the rhombus symbol shows the worldwide prevalence for all studies (Fig. 3 ). The highest prevalence was reported in Salvado et al. [ 61 ]; 3463 per 100,000 population and the lowest prevalence Bettini et al. [ 58 ]; 0.006 people reported per 100,000 population.

Funnel plot Results for estimating the prevalence of Myasthenia Gravis worldwide

Estimation of the prevalence of Myasthenia Gravis in the world based on a random-effects model
According to different reports of MG prevalence in different parts of the world, subgroup analysis by different continents (Asia, Europe, Africa and America) is reported in Table 2 , which has the highest prevalence in the Americas with 19 people (95% CI 15–23.8) (Table 2 ).
The analytical part of the study
Summary of how to enter articles: In the first stage, 4672 articles (4596 articles in international databases, 45 articles in Persian databases and 31 studies in reviewing the sources of articles) were found, and 3126 studies that were repeated in different databases were deleted. 1546 studies were entered in the screening stage, and based on the inclusion and exclusion criteria, the article was removed by reviewing the title and abstract of the 1992 studies. In the next stage (competency assessment), out of the remaining 175 studies from the screening stage, 183 articles were removed by reviewing the full text of the article because it was not relevant to the research. The remaining 22 articles were evaluated qualitatively by the CONSORT checklist, of which 2 studies were of low quality according to the criteria of this tool and were excluded from the study. Therefore, 20 articles related to the analytical part of the study were included in the systematic review and meta-analysis process (Fig. 4 ).

Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA 2009) flow diagram Analytical section
General characteristics of analytical studies
The total sample size was 643 in the drug group and 619 in the placebo group. The studies were published between 1976 and November 15, 2020. The smallest sample size was related to the study of Benatar et al. [ 109 ] with 6 patients in the drug group and 5 patients in the placebo group, and the largest sample size was related to the study of Sanders et al. [ 112 ] with 88 patients in the drug group and 88 patients in the placebo group. Summary of study characteristics including the name of the first author, year of publication, place of study, sample size, type of drug, and mean and standard deviation before and after the intervention of QMGS, Anti-AchR antibodies, SFEMG and MG-ADL indices are reported in Table 3 .
Immunoglobulin or plasma exchange drugs
A total of 13 studies examined the effect of immunoglobulin or plasma exchange drugs on MG patients. Studies were reported from 1997 to 2020. 11 studies examined the QMGS index, 4 studies the Anti-AChR antibodies, 4 studies the SFEMG index, and 3 the MG-ADL index.
MG-ADL index
The Daily Living Activity Scale (MG-ADL) is an 8-item scale to assess secondary ocular disability (two items), bulbar (three items), respiratory (one item), limb (two items) related to myasthenia gravis effects. This scale has a linear rating from zero to 3, and its overall scoring range is from zero to 24 [ 115 ]. The patient's MG-ADL questionnaire is completely reported without training and specialized equipment and usually lasts less than five minutes [ 116 ].
Based on the present meta-analysis results between studies, there is a lot of heterogeneity (I 2 = 78.5), so the stochastic effects model was used to combine the studies and the final result. Begg and Mazumdar rank correlation to test the publication bias in the studies for the MG-ADL index (P = 1.000) (Table 4 ).
As a result of the combination of studies, the mean score of MG-ADL indices before the intervention in the drug group was 8.2 ± 1.3. After the intervention was 4.0 ± 4.84 (Table 4 ), as well as the difference between the mean of the MG-ADL index before and after the intervention, 1.3 ± 0.63 was obtained (P < 0.01) (Fig. 5 ), which indicates the positive effect of Immunoglobulin or plasma exchange on the reduction of MG-ADL index.

Accumulation chart of studies included in the meta-analysis based on the difference between the mean MG-ADL index before and after the intervention for Immunoglobulin or plasma exchange
SFEMG index
Single-fiber electromyography (SFEMG) is an efficient tool to investigate neurotransmitter disorders. In this method, with the help of bipolar needle electrodes, the action potential of two adjacent muscle fibres belonging to a motor unit that have been activated voluntarily or electrically stimulated can be recorded [ 117 ]. This technique is more time consuming than conventional EMG, and patient cooperation in this method is effective because even small movements by the patient can lead to loss or change of action potential [ 118 ].
Based on the present meta-analysis results between studies, there is a lot of heterogeneity (I 2 = 43.5), so the stochastic effects model was used to combine the studies and the final result. Begg and Mazumdar rank correlation tests were not available in studies for the SFEMG index (P = 1.000) (Table 4 ). As a result of the combination of studies, the mean score of SFEMG indices before the intervention in the drug group was 82.2 ± 1.43, and after the intervention was 54/5 ± 27/5 (Table 4 ), also, the difference between the mean of SFEMG index before and after the intervention was 1.5 ± 0.73 (P < 0.01) (Fig. 6 ), which indicates the positive effect of immunoglobulin or plasma exchange on the reduction of SFEMG index.

Accumulation chart of studies included in the meta-analysis based on the difference between the mean SFEMG index before and after the intervention for Immunoglobulin or plasma exchange
Anti-AChR antibodies index
Weakness and fatigue in myasthenia gravis are caused by a decrease in acetylcholine receptors due to an autoimmune attack of antibodies at the neuromuscular junction. Specific antibodies induce this autoimmune response against the acetylcholine receptor by blocking or binding to the receptor or postsynaptic membrane damage [ 119 ].
Based on the present meta-analysis results, there is a lot of heterogeneity between studies (I 2 = 99.8), so the stochastic effects model was used to combine the studies and the final result. Begg and Mazumdar rank correlation test Emission bias was not presented in studies for Anti-AChR antibodies index (P = 1.000) (Table 4 ). As a result of the combination of studies, the mean score of Anti-AChR antibodies before the intervention in the drug group was 10.8 ± 4.6 and after the intervention was 52.7 ± 34.1 (Table 4 ). AChR antibodies were obtained before and after the intervention at − 2.006 ± 78.7 (P < 0.01) (Fig. 7 ), indicating that Immunoglobulin or plasma exchange did not affect the Anti-AChR antibodies index. QMGS index.

Accumulation chart of studies included in the meta-analysis based on the mean difference of the anti-AChR antibodies index before and after the intervention for Immunoglobulin or plasma exchange
Myasthenia Gravis Quantitative Score (QMGS) is a 13-item scale developed by Tindall et al. [ 120 ] and modified by Barohn et al. [ 121 ] to be used to determine the severity of myasthenia gravis. This scale measures ocular, bulbar, respiratory, and limb function and scores each finding from zero (no myasthenic findings) to 39 (maximum myasthenic defects) [ 122 , 123 ].
Based on the present meta-analysis results, there is a lot of heterogeneity between studies (I 2 = 98.6), so the random-effects model was used to combine the studies and the final result. Begg and Mazumdar rank correlation test was not possible in the studies for the QMGS index (P = 0.391) (Table 4 ).
As a result of the combination of studies, the mean score of QMGS indices before the intervention in the drug group was 11.2 ± 1.6 and after the intervention was 9/1 ± 4/8 (Table 4 ), as well as the difference between the mean of the QMGS index before and after the intervention. 0.62 ± 0.28 was obtained (P < 0.01) (Fig. 8 ), which indicates the positive effect of Immunoglobulin or plasma exchange on QMGS index reduction.

Accumulation diagram of meta-analysis studies based on mean differences in mean QMGS before and after intervention for Immunoglobulin or plasma exchange
Mycophenolate
In total, 4 studies examined the effect of Mycophenolate on MG patients. 4 studies reviewed the QMGS index, 2 studies the Anti-AChR antibodies index, 1 study the SFEMG index and 3 studies the MG-ADL index.
Based on the results of the present meta-analysis studies, there is a lot of heterogeneity (I 2 = 85.3), so the stochastic effects model was used to combine the studies and the outcome. Begg and Mazumdar rank correlation test of publication bias was not possible in the studies for MG-ADL index (P = 1.000) (Table 4 ).
As a result of the combination of studies, the mean score of MG-ADL indices before the intervention in the drug group was 5.9 ± 0.87 and after the intervention was 7.4 ± 5.09 (Table 4 ), as well as the difference between the mean of the MGADL index before and after The intervention showed 1.4 ± 0.9 (P < 0.01) (Fig. 9 ) which indicates the positive effect of Mycophenolate on the reduction of MG-ADL index.

Accumulation chart of studies entered for meta-analysis based on the difference between the mean MG-ADL index before and after intervention for Mycophenolate
Based on the present meta-analysis results between studies, according to a study, there was no heterogeneity (I 2 = 0), so the fixed effects model was used to combine the study and the final result. It was not possible to perform Begg and Mazumdar rank correlation test in the studies for the SFEMG index according to the study of only one research (Table 4 ).
As a result of the combination of studies, the mean score of SFEMG indices before the intervention in the drug group was 71.5 ± 11.3 and after the intervention was 60.5 ± 13.1 (Table 4 ), as well as the difference between the mean of the SFEMG index before and after the intervention. 0.9 ± 0.56 was obtained (P < 0.01), indicating Mycophenolate's positive effect on SFEMG index reduction.
Index of anti-AChR antibodies
Based on the present meta-analysis results between studies, there is a lot of heterogeneity (I 2 = 79.1), so the stochastic effects model was used to combine the studies and the final result. It was not possible to perform Begg and Mazumdar rank correlation test, publication bias in studies for Anti AChR antibodies index due to review of only 2 studies (Table 4 ).
As a result of the combination of studies, the mean score of Anti-AChR antibodies before the intervention in the drug group was 11.1 ± 2.1 and after the intervention was 5.5 ± 2.4 (Table 4 ) and the difference between the mean of the anti-AChR index. Antibodies were obtained before and after the intervention (1.9 ± 1.5 (P < 0.01) (Fig. 10 )), which indicates the positive effect of Mycophenolate on the Anti-AChR antibodies index.

Accumulation chart of studies included in the meta-analysis based on the mean difference of the anti-AChR antibodies index before and after the intervention for Mycophenolate
Based on the present meta-analysis results between studies, there is a lot of heterogeneity (I 2 = 67.9), so the stochastic effects model was used to combine the studies and the final results.
As a result of the combination of studies, the mean score of QMGS indices before the intervention in the drug group was 12.3 ± 0.71 and after the intervention was 8.0 ± 0.59 (Table 4 ). It was obtained 1.4 ± 0.77 (P < 0.01) (Fig. 11 ), indicating Mycophenolate's positive effect on QMGS index reduction.

Accumulation chart of studies entered for meta-analysis based on the mean difference of QMGS index before and after intervention for Mycophenolate
Corticosteroids
In the study of studies in the field of corticosteroids, only the QMGS index could be examined. Based on this, 3 studies examined the effect of corticosteroids on MG patients. In the study of Benatar et al. [ 109 ], the QMGS index before the intervention in the placebo group was 6.5 ± 1.8 units and in the drug group was 6.0 ± 0.2 units, and after the intervention in the placebo group decreased by 0.05 units (P > 0.05). There was a significant decrease of 2.25 units (P < 0.05) [ 109 ]. Also, in the study of Howard et al. [ 110 ] QMGS index before the intervention in the placebo group was 7.97 ± 0.91 units and in the drug group was 8.18 ± 0.63 units and after the intervention in the placebo group increased by 0.07 units (P > 0.05) and in the drug group had a decrease of 0.03 units (P > 0.05) [ 110 ]. In the study of Lindberg et al. [ 111 ], The anti-AChR antibodies index was reported before intervention in the placebo group of 264 ± 401 (μmol/L) and in the drug group of 354 m 290 (μmol/L) [ 111 ].
Based on the present meta-analysis results between studies, there is a lot of heterogeneity (I 2 = 99.4), so the stochastic effects model was used to combine the studies and the final result of the outcomes. According to the review of only two studies, it was not possible to use the Begg and Mazumdar rank correlation test for the QMGS index studies according to the review of only 2 studies (Table 4 ).
As a result of the combination of studies, the mean score of QMGS indices before the intervention in the drug group was 7.08 09 1.09 and after the intervention was 5.9 2 2.2 (Table 4 ), as well as the difference between the mean scores of the QMGS index before and after the intervention. 1.64 1 1.6 was obtained (P.010.01) (Fig. 12 ), which indicates the positive effect of corticosteroids on reducing the QMGS index.

Accumulation diagram of studies included in the meta-analysis based on the difference between the mean QMGS index before and after the intervention for Corticosteroids
Myasthenia gravis (MG) is the largest group of neuromuscular disorders caused by autoimmune antibodies against postsynaptic components of the voluntary muscle endplate [ 124 , 125 , 126 ]. Acetylcholine receptor antibodies (AChR), muscle-specific kinase (MuSK), and lipoprotein-associated protein (LRP4) have been well established as sensitive diagnostic markers and pathogens, in addition to antibodies in the classification of patients with Myasthenia gravis also play a key role [ 127 ].
Although the clinical features of MG can vary, increasing muscle weakness with continued skeletal muscle activity is one way to diagnose the disease [ 128 ]. Unlike ocular involvement, which is often asymmetric and involves several muscles, the pattern of muscle involvement in myasthenia gravis is usually symmetrical. Muscle weakness usually increases with exercise and frequent muscle use, and its intensity varies from day to day and fluctuates throughout the day [ 129 ].
In the current systematic review and meta-analysis study, the overall prevalence of MG in the world; 12.4 people per 100,000 population were obtained. Most prevalent in Salvado et al. [ 61 ]; 3463 per 100,000 population and the lowest prevalence Bettini et al. [ 58 ]; 0.006 people per 100,000 population reported.
Due to the different reports of MG prevalence in different parts of the world, a detailed study of the prevalence of this disease in different continents in order to pay more attention to planners and its consequences seemed necessary. Therefore, according to the subgroup analysis by different continents (Asia, Europe, Africa, and America), the highest prevalence of myasthenigraphy was reported in the Americas with 19 per 100,000 people and the lowest in continental Europe with 10 per 100,000 people.
Symptomatic, safe, and supportive approaches are very effective in treating myasthenia gravis, and treatment should be aimed at complete or almost complete drug recovery [ 130 ]. Most patients with myasthenia gravis to achieve therapeutic goals of full physical function or relatively high quality of life need immunosuppressive drugs. Immunosuppressive drugs are prescribed to all patients who respond only to symptomatic and supportive treatment [ 131 ].
Only the QMGS index could be assessed in studies of corticosteroids, which measures the severity of myasthenia gravis in 13 items [ 120 ]. The mean score of the QMGS index before and after the intervention in the drug group was 7.08 09 1.09 and 5.9 2 2.2, which indicates the positive effect of corticosteroid use on reducing the QMGS index improving myasthenia gravis.
Oral corticosteroid therapy has been used since the 1950s with a dramatic improvement in approximately 70 to 80% of patients with myasthenia gravis [ 132 , 133 ]. The usefulness of oral steroids is determined by the occurrence of a wide range of dose and time-dependent side effects [ 134 , 135 ]. Intermittent intravenous methylprednisolone (IVMP) is used to treat several autoimmune disorders, including MG, on the assumption that it is more effective and has fewer side effects than oral steroids [ 136 ]. IVMP is also effective in severe cases of MG [ 137 ].
Mycophenolate mofetil (MMF) is an immunosuppressive agent that is primarily used to prevent acute rejection of organ transplants [ 138 ] which have reported preliminary use of this drug in the treatment of myasthenia gravis [ 139 ].
Regarding the effectiveness of mycophenolate mofetil, the mean score of MG-ADL index before and after the intervention in the drug group was 5.9 87 0.87 and 7.4 09 5.09, respectively. This scale assesses daily life activity in people with myasthenia gravis through 8 items [ 115 ]. The mean score of the SFEMG (single-strand electromyography) index, which is used to evaluate neuromuscular site abnormalities [ 117 ], was reported to be 71.5 ± 11.3 and 60.13 5 5.1, respectively, before and after the intervention in the drug group. Also, the mean score of Anti-AChR antibodies before and after the intervention in the drug group was 5/5 ± 2/4 and 11/2 ± 1/1. The mean score of QMGS indices before and after the intervention in the drug group was 12.3 ± 0.71 and 8.0 ± 1.59, which the results show the positive effect of using Mycophenolate on reducing the above 4 indicators and thus improving the treatment status of patients with MG.
Certain cure requires suppression or modulation of the immune system by intravenous immunoglobulin (IVIg) or plasma replacement (PLEX) [ 140 ]. Immune system modification is used when rapid recovery is required, such as exacerbated myasthenia gravis, power optimization before thymectomy, and patients who do not tolerate and respond adequately to immunosuppressive drugs [ 100 , 141 , 142 ]. In recent years, the administration of 2 g/kg intravenous IVIg immunoglobulin has been proven to treat moderate to severe myasthenia gravis and is continuously used to manage intensified MG [ 143 ].
Therapeutic plasmapheresis or plasma replacement (PLEX) is the first line of treatment in patients with myasthenia gravis with respiratory failure, inability to swallow, myasthenic crisis, or inadequate response to drug therapy [ 144 , 145 ]. In therapeutic plasmapheresis, plasma containing pathogenic antibodies is separated from the patient's blood and returned to other cells. Plasma replacement is prescribed five times in 10 to 14 days, through which and by repeating it, plasma levels of acetylcholine receptor antibody are reduced, and clinical improvement is achieved [ 119 ].
Due to the high prevalence of myasthenia gravis globally and its many negative consequences for individuals and society. Therefore, it seems useful to take measures to achieve better therapies or to use supportive therapies to reduce the symptoms of the disease. Common drug treatments in MG were evaluated to show the effectiveness of immunosuppressive drugs, including steroids and their modulators, including intravenous immunoglobulin (IVIg and plasma replacement) (PLEX). These studies can provide useful information to health care providers, enrich health care interventions, improve the quality of services, and ultimately improve the quality of life of these people. Therefore, it is suggested that physicians and the health care system give these drug classes more attention.
The application of nanotechnology is promising, given frustrating problems in therapeutic neurology [ 146 ]. Nanotechnology involves the manipulation of technological machinery at the atomic scale. For perspective, a nucleus is about 6 μm across, a ribosome 20 nm in diameter, and a single strand of DNA 2 nm wide [ 146 ]. A typical human being is composed of 100 trillion cells. Nanotechnology has created novel devices for the treatment of various neurological diseases. Shrinkage of machinery, chip-based technologies, and the creation of unprecedented nanomaterials are contributing immensely to the reduction of morbidity [ 146 , 147 ]
Considerable efforts are being focused on using nanoneuromedicine for disease treatment in the research laboratory. In the case of neurodegenerative diseases such as myasthenia gravis (MG), Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS), nanomedicines have emerged as promising treatment options. Pathophysiological processes involving neuron inflammation and protein misfolding initiate a degeneration cycle within the cell. This can be thwarted using better drug targeting. Diagnosing and monitoring the end-effects of therapeutics is possible using nanoneurotechnology [ 146 , 147 , 148 ].
In addition to what has been said, the interest in graphene-based nanomaterials (GBNs) application in nanomedicine, particularly neurology, steadily increased in the last decades. GBNs peculiar physical–chemical properties allow the design of innovative therapeutic tools to manipulate biological structures with subcellular resolution [ 148 , 149 ]. Based on the study, it can also be said that to develop effective antioxidant therapies the best strategy may be to create new nanoscale drug delivery systems [ 150 ].
Limitations
Among the limitations of this study, it can be asserted that some samples were not based on random selection. Also, non-uniform reporting of articles, inconsistent implementation method, non-copying and unavailability of the full text of articles presented at the conference can be mentioned as other limitations.
Suggestion for future works
The meta-analysis results enable the overall prevalence to be presented to the policy-maker and thus manage the cost, time and future diagnostic and treatment decisions commensurate with the overall prevalence. A systematic review also reveals drugs effective in treating myasthenia gravis, which can guide physicians and encourage the researcher to conduct future clinical trial studies and a network meta-analysis to determine therapeutic supplements for the disease.
The results of systematic review of drug evaluation in patients with myasthenia gravis showed that Mycophenolate and Immunoglobulin or plasma exchange drugs have positive effects in the treatment of MG. It also represents the positive effect of immunoglobulin or plasma exchange on reducing SFEMG index and QMGS index and the positive effect of Mycophenolate on reducing MG-ADL index, SFEMG and Anti-AChR antibodies index. In addition to what was mentioned, based on a meta-analysis of the random-effect model, the overall prevalence of MG in the world is 12.4 people per 100,000 populations, which indicates the urgent need for the attention of officials and specialists to this disease for prevention and treatment.
Availability of data and materials
Datasets are available through the corresponding author upon reasonable request.
Abbreviations
Scientific Information Database
Medical Subject Headings
Web of science
Preferred Reporting Items for Systematic Reviews and Meta-Analysis
Consolidated Standards of Reporting Trials
Strengthening the reporting of observational studies in epidemiology for a cross-sectional study
Myasthenia Gravis
Anti-acetylcholine receptor antibodies
Intravenous immunoglobulin
Quantitative Myasthenia Gravis Score
Myasthenia Gravis Activities of Daily Living
Single-fibre electromyography
Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87(4):419–25.
PubMed PubMed Central Google Scholar
Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023–36.
CAS PubMed Google Scholar
Shield TW, editor. General thoracic surgery, 7th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2010. pp. 2323–64.
Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116(11):2843–54.
CAS PubMed PubMed Central Google Scholar
Benatar M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. 2006;16(7):459–67.
PubMed Google Scholar
Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B. Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun. 2014;48–49:143–8.
Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8(5):475–90.
Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7(3):365–8.
Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. 2011;69(2):418–22.
Kerty E, Elsais A, Argov Z, Evoli A, Gilhus NE. EFNS/ENS guidelines for the treatment of ocular myasthenia. Eur J Neurol. 2014;21:687–93.
Heldal AT, Owe JF, Gilhus NE, Romi F. Seropositive myasthenia gravis: a nationwide epidemiologic study. Neurology. 2009;73:150–1.
Wang L, Zhang S, Xi J, Li W, Zhou L, Lu J, et al. Efficacy and safety of tacrolimus for myasthenia gravis: a systematic review and meta-analysis. J Neurol. 2017;264(11):2191–200.
Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population-based epidemiological studies in myasthenia Gra-vis. BMC Neurol. 2010;10:46.
MacDonald BK, Cockerell OC, Sander JW, Shorvon SD. The incidence and lifetime prevalence of neurological disorders in a prospective community-based study in the UK. Brain J Neurol. 2000;123(Pt 4):665–76.
Google Scholar
Breiner A, Widdifield J, Katzberg HD, Barnett C, Bril V, Tu K. Epidemiology of myasthenia gravis in Ontario, Canada. Neuromuscular Disord. 2016;26(1):41–6.
Kurtzke JF. Epidemiology of myasthenia gravis. Adv Neurol. 1978;19:545–66.
Eaton WW, Rose NR, Kalaydjian A, Pedersen MG, Mortensen PB. Epidemiology of autoimmune diseases in Denmark. J Autoimmun. 2007;29(1):1–9.
Mh H, Ms F. The clinical outcome of thymectomy in myasthenia gravis. Tehran Univ Med J TUMS Publications. 2009;66(11):821–5.
Chen S, Xu M-B, Zhou X-L, Rong P-Q, Jin T-Y, Zheng G-Q. Chinese herbal medicine for myasthenia gravis: a systematic review and meta-analysis. Front Pharmacol. 2018;9:969.
Li H-F, Xie Y, Yue Y-X. Myasthenia gravis: subgroup classifications. Lancet Neurol. 2016;15(4):355–6.
Meriggioli MN, Rowin J, Richman JG, Leurgans S. Mycophenolate mofetil for myasthenia gravis: a double-blind, placebo-controlled study. Ann NY Acad Sci. 2003;998:494–9.
Sieb J. Myasthenia gravis: an update for the clinician. Clin Exp Immunol. 2014;175(3):408–18.
De Feo LG, Schottlender J, Martelli NA, Molfino NA. Use of intravenous pulsed cyclophosphamide in severe, generalized myasthenia gravies. Muscle Nerve. 2002;26(1):31–6.
García-Carrasco M, Escárcega RO, Fuentes-Alexandro S, Riebeling C, Cervera R. Therapeutic options in autoimmune myasthenia gravis. Autoimmun Rev. 2007;6(6):373–8.
Ciafaloni E. Mycophenolate mofetil and myasthenia gravis. Lupus. 2005;14(3-suppl):46–9.
Skeie GO, Apostolski S, Evoli A, et al. Guidelines for the treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2006;13(7):691–9.
Rozsa C, Lovas G, Fornadi L, Szabo G, Komoly S. Safety of long-term combined immunosuppressive treatment in myasthenia gravis: analysis of adverse effects of 163 patients. Eur J Neurol. 2006;13(9):947–52.
Drachman DB, Adams RN, Hu R, et al. Rebooting the immune system with high-dose cyclophosphamide for the treatment of refractory myasthenia gravis. Ann N Y Acad Sci. 2008;1132:305–14.
Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med. 2013;86(2):255–60.
Gajdos P, Tranchant C, Clair B, Bolgert F, Eymard B, Stojkovic T, et al. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol. 2005;62(11):1689–93.
Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIgand PLEX in patients with myasthenia gravis. Neurology. 2011;76(23):2017–23.
Katzberg HD, Barnett C, Bril V. Predictors of response to immunomodulation in patients with myasthenia gravis. Muscle Nerve. 2012;45(5):648–52.
Nagayasu T, Yamayoshi T, Matsumoto K, Ide N, Hasazumi S, Nomura M, et al. Beneficial effects of plasmapheresis before thymectomy on the outcome in myasthenia gravis. Jpn J Thorac Cardiovasc Surg. 2005;53(1):2–7.
Guptill JT, Sharma BK, Marano A, Soucy A, Krueger A, Sanders DB. The estimated cost of treating myasthenia gravis in an insured US population. Muscle Nerve. 2012;45(3):363–6.
Henderson LK, Craig JC, Willis NS, Tovey D, Webster AC. How to write a cochrane systematic review. Nephrology. 2010;15(6):617–24.
Ramke J, Palagyi A, Jordan V, Petkovic J, Gilbert CE. Using the STROBE statement to assess reporting in blindness prevalence surveys in low and middle-income countries. PloS One. 2017;12(5):e0176178.
Schulz KF, Altman DG, Moher D, Group C. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Trials. 2010;11(1):32.
Murai H, Yamashita N, Watanabe M, Nomura Y, Motomura M, Yoshikawa H, et al. Characteristics of myasthenia gravis according to onset-age: Japanese nationwide survey. J Neurol Sci. 2011;305(1–2):97–102.
Nemet AY, Kaiserman I, Mimouni M, Segal O, Vinker S. High prevalence of myasthenia gravis among rural adult populations. J Clin Neuromuscul Dis. 2014;16(2):47–50.
Park S-Y, Lee JY, Lim NG, Hong Y-H. Incidence and prevalence of myasthenia gravis in Korea: a population-based study using the National Health Insurance claims database. J Clin Neurol. 2016;12(3):340–4.
Lee HS, Lee HS, Shin HY, Choi Y-C, Kim SM. The epidemiology of myasthenia gravis in Korea. Yonsei Med J. 2016;57(2):419–25.
Okinaka S, Reese HH, Katsuki S, et al. The prevalence of multiple sclerosis and other neurological diseases in Japan. Acta Neurologica Scandinavica. 1966;47(Suppl 19):68–76.
Araki S, Uchino M, Yoshida O. Epidemiologic study of multiple sclerosis, myasthenia gravis and polymyositis in the city of Kumamoto, Japan. Clin Neurol. 1983;23:838–41.
CAS Google Scholar
Kondo K, Takasu T, Ahmed A. Neurological diseases in Karachi, Pakistan—elevated occurrence of subacute sclerosing panencephalitis. Neuroepidemiology. 1988;7:66–80.
Yu YL, Hawkins BR, Ip MS, Wong V, Woo E. Myasthenia gravis in Hong Kong Chinese: epidemiology and adult disease. Acta Neurol Scand. 1992;86(2):113–9.
Zieda A, Ravina K, Glazere I, Pelcere L, Naudina M, Liepina L, et al. A nationwide epidemiological study of myasthenia gravis in Latvia. Eur J Neurol. 2018;25(3):519–26.
Lavrnic D, Basta I, Rakocevic-Stojanovic V, Stevic Z, Peric S, Nikolic A, et al. Epidemiological study of adult-onset myasthenia gravis in the area of Belgrade (Serbia) in the period 1979–2008. Neuroepidemiology. 2013;40(3):190–4.
Tola M, Granieri E, Paolino E, Caniatti L, Quatrale R, Mazzanti B, et al. Epidemiological study of myasthenia gravis in the province of Ferrara, Italy. J Neurol. 1989;236(7):388–90.
Montomoli C, Citterio A, Piccolo G, Cioccale R, Ferretti VV, Fratti C, et al. Epidemiology and geographical variation of myasthenia gravis in the province of Pavia, Italy. Neuroepidemiology. 2012;38(2):100–5.
Cetin H, Fülöp G, Zach H, Auff E, Zimprich F. Epidemiology of myasthenia gravis in Austria: rising prevalence in an ageing society. Wien Klin Wochenschr. 2012;124(21–22):763–8.
Storm-Mathisen A. Epidemiology of myasthenia gravis in Norway. Acta Neurol Scand. 1984;70(4):274–84.
Westerberg E, Punga AR. Epidemiology of Myasthenia gravis in Sweden 2006–2016. Brain Behav. 2020;10:e01819.
Kalb B, Matell G, Pirskanen R, Lambe M. Epidemiology of myasthenia gravis: a population-based study in Stockholm, Sweden. Neuroepidemiology. 2002;21(5):221–5.
Aiello I, Pastorino M, Sotgiu S, Pirastru M, Sau G, Sanna G, et al. Epidemiology of myasthenia gravis in northwestern Sardinia. Neuroepidemiology. 1997;16(4):199–206.
Guidetti D, Sabadini R, Cavalletti S, Lodesani M, Mantegazza R, Solime FCV. Epidemiological study of myasthenia gravis in the province of Reggio Emilia, Italy. Eur J Epidemiol. 1998;14(4):381–7.
Foldvari A, Kovacs N, Sipos V, Merth G, Vincze F, Szucs M, et al. Estimation of incidence, prevalence, and age-at-diagnosis of myasthenia gravis among adults by hospital discharge records. Wien Klin Wochenschr. 2015;127(11–12):459–64.
Zivadinov R, Jurjevic A, Willheim K, Cazzato G, Zorzon M. Incidence and prevalence of myasthenia gravis in the County of the Coast and Gorski kotar, Croatia, 1976 through 19961. Neuroepidemiology. 1998;17(5):265–72.
Bettini M, Chaves M, Cristiano E, Pagotto V, Perez L, Giunta D, et al. Incidence of autoimmune myasthenia gravis in a health maintenance organization in Buenos Aires, Argentina. Neuroepidemiology. 2017;48(3–4):119–23.
Andersen J, Heldal A, Engeland A, Gilhus N. Myasthenia gravis epidemiology in a national cohort; combining multiple disease registries. Acta Neurol Scand. 2014;129:26–31.
Aragones J, Altimiras J, Roura P, Alonso F, Bufill E, Munmany A, et al. Prevalence of myasthenia gravis in the Catalan county of Osona. Neurología (English Edition). 2017;32(1):1–5.
Salvado M, Canela M, Ponseti JM, Lorenzo L, Garcia C, Cazorla S, et al. Study of the prevalence of familial autoimmune myasthenia gravis in a Spanish cohort. J Neurol Sci. 2016;360:110–4.
Christensen P, Jensen T, Tsiropoulos I, Søsrensen T, Kjser M, Højer-Pedersen E, et al. Incidence and prevalence of myasthenia gravis in western Denmark: 1975 to 1989. Neurology. 1993;43(9):1779.
Robertson N, Deans J, Compston D. Myasthenia gravis: a population-based epidemiological study in Cambridgeshire, England. J Neurol Neurosurg Psychiatry. 1998;65(4):492–6.
Garland H, Clark ANG. Myasthenia gravis, a personal study of 60 cases. BMJ. 1956;1:1259–62.
Pennington GW, Wilson A. Incidence of myasthenia gravis in the Merseyside conurbation. In: Veits HR, editor. Myasthenia Gravis. Proceedings of the second international symposium. Springfield IL: Charles C Thomas; 1961; pp. 337–45.
Gudmundsson KR. The prevalence of some neurological diseases in Iceland. Acta Neurol Scand. 1968;44:55–69.
Oosterhuis: Epidemiologie dei myasthenie in Amsterdam. Neurologie Deutsche Jesells. 1977; 103–108.
Hokkanen E. Epidemiology of myasthenia gravis in Finland. J Neurol Sci. 1969;9:463–78.
Giagheddu M, Puggioni G, Sanna G, et al. Epidemiological study of myasthenia gravis in Sardinia Italy (1958–1986). Acta Neurol Scand. 1989;79:326–33.
D’Alessamdro R, Granieri E, Benassi G, et al. Comparative study on the prevalence of myasthenia gravis in the provinces of Bologna and Ferrera Italy. Acta Neurol Scand. 1991;83(2):83–8.
Sorensen TT, Holm EB. Myasthenia gravis in the county of Viborg, Denmark. Eur Neurol. 1989;29:177–9.
Somnier FE, Keiding N, Paulson OB. Epidemiology of myasthenia gravis in Denmark: a longitudinal and comprehensive population survey. Arch Neurol. 1991;48(7):733–9.
Christensen PB, Jensen TS, Tsiropoulos I, et al. Mortality and survival in myasthenia gravis: a Danish population-based study. JNNP. 1998;64(1):78–83.
Ferrari G, Lovaste MG. Epidemiology of myasthenia gravis in the province of Trento (northern Italy). Neuroepidemiology. 1992;11(3):135–42.
Krivopusk ME. Clinico-epidemiological aspects of hereditary neuromuscular diseases in the Krasnodar territory. Zhurnal Nevropatologii I Psikhiatrii Imeni SS Korsakova. 1991;91(9):3–5.
Lavrnic D, Jarebinski M, Rakocevic-Stojanovic V, et al. Epidemiological and clinical characteristics of myasthenia gravis in Belgrade, Yugoslavia (1983–1992). Acta Neurologica Scandanavica. 1999;100(3):168–74.
Kyriallis K, Hristova A, Middleton I. What is the real epidemiology of myasthenia gravis? Neurology. 1995; A351.
Holtsema H, Mourik J, Rico RE, et al. Myasthenia gravis on the Dutch Antilles: an epidemiological study. Clin Neurol Neurosurg. 2000;102(4):195–8.
Villagra-Cocco A, Villagra-Cocco P. Prevalence of myasthenia gravis on the island of La Palma. Revista Neurol. 1997;25(148):2068–9.
Oopik M, Kaasik AE, Jakobson J. A population-based epidemiological study of myasthenia gravis in Estonia. JNNP. 2003;74(12):1638–43.
Wirtz PW, Nijnuis MG, Sotodeh M, et al. The epidemiology of myasthenia gravis, Lambert Eaton myasthenic syndrome and their associated tumours in the northern part of Southern Holland. J Neurol. 2003;250(6):698–701.
Kotov SV, Neretin VI, Agafonov BV, Sidorova OP. Population-based study of Myasthenia in Moscow region. Zhurnal Nevrologii I Psikhiatrii Imeni SS Korsakova. 2006;106(5):52–5.
Somnier FE. Increasing incidence of late-onset anti-AChR antibody-seropositive myasthenia gravis. Neurology. 2005;65:928–30.
Poulas K, Tsibri E, Kokla A, et al. Epidemiology of seropositive myasthenia gravis in Greece. JNNP. 2001;71(3):352–6.
Niks EH, Kuks JB, Verschuuren JJ, et al. Epidemiology of myasthenia gravis with anti-muscle specific kinase antibodies in The Netherlands. JNNP. 2007;78(4):417–8.
Tsiamalos P, Kordas G, Kokla A, et al. Epidemiological and immunological profile of muscle-specific kinase myasthenia gravis in Greece. Eu J Neurol. 2009;16(8):925–30.
Maharaj J, Bahadursingh S, Ramcharan K. Myasthenia gravis in South Trinidad. West Indian Med J. 2013;62(6):510–4.
Gordon B, Noone J, Van Doren B, Zacherle E, Blanchette C. Prevalence and cost of myasthenia gravis in the medicare beneficiary sample. Value Health. 2015;18(7):A661.
Phillips LH, Torner JC, Anderson MS, Cox GM. The epidemiology of myasthenia gravis in central and western Virginia. Neurology. 1992;42(10):1888.
Kurland LT. Descriptive epidemiology if selected neurologic and myopathic disorders with particular reference to a survey in Rochester, Minnesota. J Chronic Disorders. 1958;8(4):378.
Alter M, Rhett-Talbert O, Kurland LT. Myasthenia gravis in a southern community. Arch Neurol. 1960;3:65–9.
Kvirkveliia NB. Clinico-epidemiologic aspects of Myasthenia in the Georgian SSR. Zh Nevropatol Psikhiatr Im S S Korsakova. 1986;86(3):327–30.
Cisernos AD, Luis RS, Leon R, Carrera PL. Some epidemiological aspects of myasthenia gravis in Cuba. Revista de Neurol. 1996;24(128):435–9.
Sanchez JL, Uribe CS, Franco AF, Jimeniz ME, Arcos-Burgos OM, Palacio LG. Prevalence of myasthenia gravis in Antioquia, Colombia. Revista de Neurologia. 2002;34(11):1010–2.
Deffeminis Rospide HA, Petra de Mirabel M, Piazza de Silva N, et al. Estudio epidemiologico de la miastenia en el Uruguay. Acta Neurol Latinoamer 1975; 53–65.
Khedr EM, Fawi G, Abbas MA-A, El-Fetoh AN, Zaki AF, Gamea A, et al. Prevalence of neuromuscular disorders in Qena governorate/Egypt: a population-based survey. Neurol Res. 2016; 38(12):1056–63
El-Tallawy HN, Khedr EM, Qayed MH, Helliwell TR, Kamel NF. Epidemiological study of neuromuscular disorders in Assuit, Egypt. Neuroepidemiology. 2005;25(4):205–11.
Gattellari M, Goumas C, Worthington J. A national epidemiological study of myasthenia gravis in Australia. Eur J Neurol. 2012;19(11):1413–20.
Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C, Group MGCS. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Ann Neurol. 1997;41(6):789–96.
Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. 2007;68(11):837–41.
Wolfe GI, Barohn RJ, Foster BM, Jackson CE, Kissel JT, Day JW, Thornton CA, Nations SP, Bryan WW, Amato AA. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve. 2002;26(4):549–52.
Gamez J, Salvadó M, Carmona F, de Nadal M, Romero L, Ruiz D, Jáuregui A, Martínez O, Pérez J, Suñé P. Intravenous immunoglobulin to prevent myasthenic crisis after thymectomy and other procedures can be omitted in patients with well-controlled myasthenia gravis. Ther Adv Neurol Disord. 2019;12:1756286419864497.
Barnett TC, Bril V, Davis AM. Performance of individual items of the quantitative myasthenia gravis score. Neuromuscul Disord. 2013;23(5):413–7.
Zinman L, Bril V. IVIG treatment for myasthenia gravis: effectiveness, limitations, and novel therapeutic strategies. Ann N Y Acad Sci. 2008;1132(1):264–70.
Katzberg HD, Barnett C, Merkies IS, Bril V. Minimal clinically important difference in myasthenia gravis: outcomes from a randomized trial. Muscle Nerve. 2014;49(5):661–5.
Jacob S, Murai H, Utsugisawa K, Nowak RJ, Wiendl H, Fujita KP, O’Brien F, Howard JF Jr. Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study. Ther Adv Neurol Disord. 2020;13:1756286420911784.
Gajdos P, Chevret S. Treatment of myasthenia gravis acute exacerbations with intravenous immunoglobulin. Ann N Y Acad Sci. 2008;1132(1):271–5.
Howard JF, Bril V, Burns TM, Mantegazza R, Bilinska M, Szczudlik A, Beydoun S. Garrido FJRDR, Piehl F, Rottoli M: Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. 2019;92(23):e2661–73.
Benatar M, Mcdermott MP, Sanders DB, Wolfe GI, Barohn RJ, Nowak RJ, Hehir M, Juel V, Katzberg H, Tawil R. Efficacy of prednisone for the treatment of ocular Myasthenia (EPITOME): a randomized, controlled trial. Muscle Nerve. 2016;53(3):363–9.
Howard FM Jr, Duane DD, Lambert EH, Daube JR. Alternate-day prednisone: preliminary report of a double-blind controlled study. Ann N Y Acad Sci. 1976;274:596–607.
Lindberg C, Andersen O, Lefvert A. Treatment of myasthenia gravis with methylprednisolone pulse: a double-blind study. Acta Neurol Scand. 1998;97(6):370–3.
Sanders D, Hart I, Mantegazza R, Shukla S, Siddiqi Z, De Baets M, Melms A, Nicolle M, Solomons N, Richman DP. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71(6):400–6.
Wolfe GI, Barohn RJ, Sanders DB, McDermott MP. Comparison of outcome measures from a trial of mycophenolate mofetil in myasthenia gravis. Muscle Nerve. 2008;38(5):1429–33.
Group MS. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71(6):394–9.
Muppidi S. The myasthenia gravis-specific activities of daily living profile. Ann N Y Acad Sci. 2012;1274(1):114–9.
Muppidi S, Wolfe GI, Conaway M, Burns TM, Composite M, Group MQS. MG-ADL: still a relevant outcome measure. Muscle Nerve. 2011;44(5):727–31.
Dumitru D, Amato A, Zwarts M, editors. Electrodiagnostic medicine, 2nd ed. Philadelphia: Hanley & belfus, INC, 2002; pp. 1148–1177.
Jabre JF, Chirico-Post J, Weiner M. Stimulation SFEMG in myasthenia gravis. Muscle Nerve. 1989;12(1):38–42.
Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J. Harrison's principles of internal medicine, 19th ed. New York: McGraw-Hill Education; 2015; pp. 2704–6.
Tindall RS, Rollins JT, Phillips JT, et al. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316:719–24.
Barohn RJ, McIntire D, Herbelin L, et al. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769–72.
Sharshar T, Chevret S, Mazighi M, et al. Validity and reliability of two muscle strength scores commonly used as endpoints in assessing treatment of myasthenia gravis. J Neurol. 2000;247:286–90.
Bedlack RS, Simmel D, Bosworth H, et al. Quantitative myasthenia gravis score: assessment of responsiveness and longitudinal validity. Neurology. 2005;64:1968–70.
Gilhus NE. Myasthenia and neuromuscular junction. Curr Opin Neurol. 2012;25:523–9.
Querol L, Illa I. Myasthenia and the neuromuscular junction. Curr Opin Neurol. 2013;26:459–65.
Verschuuren JJ, Huijbers MG, Plomp JJ, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun Rev. 2013;12:918–23.
Huijbers M, Lipka A, Plomp J, Niks E, van der Maarel S, Verschuuren J. Pathogenic immune mechanisms at the neuromuscular synapse: specific antibody-binding epitopes’ role in myasthenia gravis. J Intern Med. 2014;275(1):12–26.
Zisimopoulou P, Brenner T, Trakas N, Tzartos SJ. Serological diagnostics in myasthenia gravis based on novel assays and recently identified antigens. Autoimmun Rev. 2013;12:924–30.
Verschuuren J, Strijbos E, Vincent A. Neuromuscular junction disorders. Handb Clin Neurol. 2016;133:447–66.
Skeie GO, Apostolski S, Evoli A, et al. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2010;17:893–902.
Hart IK, Sathasivam S, Sharshar T. Immunosuppressive agents for myasthenia gravis. Cochrane Database Syst Rev. 2007;4:CD005224.
Brunner NG, Namba T, Grob D. Corticosteroids in management of severe, generalized myasthenia gravis. Effectiveness and comparison with corticotropin therapy. Neurology. 1972;22:603–10.
Mann JD, Johns TR, Campa JF, Muller WH. Long-term prednisone followed by thymectomy in myasthenia gravis. Ann NY Acad Sci. 1976;274:608–22.
Pascuzzi RM, Coslett HB, Johns TR. Long-term cortico- steroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15:291–8.
Evoli A, Batocchi AP, Palmisani MT, Monaco ML, Tonali P. Long-term results of corticosteroid therapy in patients with myasthenia gravis. Eur Neurol. 1992;32:37–43.
Matell G, Baerendtz S, Hulting J, Malmlund HO. Effects on Myasthenia of twin shock doses of methylprednisolone (TSDMP). 1982. 5th Int Congr Neuromusc Diseases.
Arsura E, Brunner NG, Namba T, Grob D. High-dose intravenous methylprednisolone in myasthenia gravis. Arch Neurol. 1985;42:1149–53.
Chaudhry V, Cornblath D, Griffin J, O’Brien R, Drachman DB. Mycophenolate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology. 2001;56(1):94–6.
Ciafaloni E, Massey J, Tucker-Lipscomb B, Sanders D. Mycophenolate mofetil for myasthenia gravis: an open-label pilot study. Neurology. 2001;56(1):97–9.
Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve. 2004;29:484–505.
Gajdos P, Chevret S, Toyka K. Plasma exchange for myas-thenia gravis. Cochrane Database Syst Rev. 2002. https://doi.org/10.1002/14651858.cd002275 .
Article PubMed PubMed Central Google Scholar
Jnsen P, Bril V. A comparison of the effectiveness of intravenous immunoglobulin and plasma exchange as preoperative therapy of myasthenia gravis. J Clin NeuromusculDis. 2008;9:352–5.
Ronager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulintreatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs. 2001;25:967–73.
Chegini A. Therapeutic plasmapheresis in myasthenic crisis after botox injection (case report). 2016 (In Persian).
McLeod BC, Weinstein R, Winters JL. Textbook of apheresis principles and practice, 3rd ed. USA: AABB; 2010. pp. 295–317.
Cellot G, Franceschi Biagioni A, Ballerini L. Nanomedicine and graphene-based materials: advanced technologies for potential treatments of diseases in the developing nervous system. Pediatr Res. 2021. https://doi.org/10.1038/s41390-021-01681-6 .
Article PubMed Google Scholar
Ambesh P, Gregory AD. Nanotechnology in neurology: genesis, current status, and prospects. Ann Indian Acad Neurol. 2015;18(4):382–6.
Sriramoju B, Kanwar RK, Kanwar JR. Nanomedicine based nanoparticles for neurological disorders. Curr Med Chem. 2014;21(36):4154–68.
Kumar Nath U, Bhattacharyya D, Chattopadhya D, Dhingra G, Azad SH, Mohanty A. Visceral leishmaniasis masquerading as drug-induced pancytopenia in myasthenia gravis. Drug Discov Therapeutics. 2021;15(1):48–50.
Eftekhari A, Maleki Dizaj S, Chodari L, Sunar S, Hasanzadeh A, Ahmadian E, Hasanzadeh M. The promising future of nano-antioxidant therapy against environmental pollutants induced-toxicities. Biomed Pharmacother. 2018;103:1018–27.
Download references
Acknowledgements
This study results from research project No. 4000257 approved by the Student Research Committee of Kermanshah University of Medical Sciences. We would like to thank the esteemed officials of the centre for the financial affords of this study.
By Deputy for Research and Technology, Kermanshah University of Medical Sciences (IR) (4000257). This deputy has no role in the study process.
Author information
Authors and affiliations.
Department of Biostatistics, School of Health, Kermanshah University of Medical Sciences, Kermanshah, Iran
Nader Salari
Student Research Committee, Kermanshah University of Medical Sciences, Kermanshah, Iran
Behnaz Fatahi & Mohsen Kazeminia
Department of Translation Studies, Faculty of Literature, Istanbul University, Istanbul, Turkey
Yalda Bartina
Department of Neurosurgery, School of Medicine, Kermanshah University of Medical Sciences, Kermanshah, Iran
Reza Fatahian
Department of Neurology, School of Medicine, Kermanshah University of Medical Sciences, Kermanshah, Iran
Payam Mohammadi
Department of Biology, Faculty of Science, University Putra Malaysia, Serdang, Selangor, Malaysia
Shamarina Shohaimi
Cellular and Molecular Research Center, Gerash University of Medical Sciences, Gerash, Iran
Masoud Mohammadi
You can also search for this author in PubMed Google Scholar
Contributions
NS, BF, and MK contributed to the design and MM statistical analysis and participated in most study steps. MM and BF, and MK prepared the manuscript. YB and RF and MM and BF and PM and SS helped design and interpret the study. All authors read and approved the content of the manuscript.
Corresponding author
Correspondence to Masoud Mohammadi .
Ethics declarations
Ethics approval and consent to participate.
Ethics approval was received from the deputy of the research and technology ethics committee, Kermanshah University of Medical Sciences (IR.KUMS.REC.1400.116).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Salari, N., Fatahi, B., Bartina, Y. et al. Global prevalence of myasthenia gravis and the effectiveness of common drugs in its treatment: a systematic review and meta-analysis. J Transl Med 19 , 516 (2021). https://doi.org/10.1186/s12967-021-03185-7
Download citation
Received : 27 October 2021
Accepted : 06 December 2021
Published : 20 December 2021
DOI : https://doi.org/10.1186/s12967-021-03185-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Myasthenia gravis
- Systematic review
- Meta-analysis
Journal of Translational Medicine
ISSN: 1479-5876
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]
Rozanolixizumab: A New Therapy in the Treatment of Myasthenia Gravis
Affiliation.
- 1 Washington State University College of Pharmacy and Pharmaceutical Sciences, Spokane, WA, USA.
- PMID: 38533739
- DOI: 10.1177/10600280241239048
Objective: The aims of this article are to review the clinical aspects of rozanolixizumab, to describe clinical trial results that led to the drug's approval, and to examine the impact on patient care to aid clinical decision making.
Data sources: A PubMed search was conducted using the terms Rystiggo ™, rozanolixizumab , rozanolixizumab therapy , and myasthenia gravis . The most recent prescribing information was also used for information relating to the drug and for identification of pertinent studies.
Study selection/data extraction: Phase I, II, and III randomized controlled trials were all eligible for inclusion. Meeting abstracts and articles focusing on the use of rozanolixizumab or any indication other than generalized myasthenia gravis were excluded from this article.
Data synthesis: Food and Drug Administration approval of rozanolixizumab is based on the phase III MycarinG study in patients with generalized myasthenia gravis. A phase II trial explored initial clinical efficacy and safety pertaining to the dose and frequency of rozanolixizumab across 2 treatment periods in patients with moderate to severe myasthenia gravis.
Relevance to patient care and clinical practice in comparison to existing agents: Rozanolixizumab is the first therapy approved to treat patients positive for both types of antibodies, anti-acetylcholine receptor or anti-muscle-specific tyrosine kinase, in generalized myasthenia gravis.
Conclusion/relevance: The approval of rozanolixizumab represents an advancement in therapy for generalized myasthenia gravis. The provision of individualized, targeted, and well-tolerated treatment is valuable for the patients whose myasthenia gravis is not well controlled and who are seeking a medication with a rapid onset of action to improve their symptoms and overall quality of life.
Keywords: generalized myasthenia gravis; immunosuppressants; myasthenia gravis; myasthenia gravis treatment; rozanolixizumab.
Publication types
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- My Account Login
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Open access
- Published: 24 January 2024
Registered trials on novel therapies for myasthenia gravis: a cross-sectional study on ClinicalTrials.gov
- Xingyue Li 1 na1 ,
- Jinxin Chen 2 na1 ,
- Youtao Wang 2 ,
- Siwei Zheng 2 ,
- Kun Wan 2 &
- Xiaodong Liu ORCID: orcid.org/0000-0001-6688-3342 3
Scientific Reports volume 14 , Article number: 2067 ( 2024 ) Cite this article
827 Accesses
Metrics details
- Neurological disorders
Novel biologics in MG therapy research is on the rise. This research aimed to investigate the characteristics of registered trials on novel therapies for myasthenia gravis on ClinicalTrials.gov. This cross-sectional study used a descriptive approach to assess the features of the included trials on ClinicalTrials.gov. We found 62 registered trials from 2007 to 2023 on ClinicalTrials.gov. The results showed a yearly rise in the number of registered trials (r = 0.76, p < 0.001). Following 2017, more industry-sponsored trials were conducted (91.5% [43] vs. 60% [9], p = 0.009), fewer results were released (10.6% [5] vs. 60% [9], p = 0.001), and more trials entered phase 3 (67.4% [31] vs. 20% [2], p = 0.001). The most researched novel medications were neonatal Fc receptor inhibitors (51.2% [21]), complement inhibitors (39.0% [16]), and B cell depletors (14.6% [6]). According to the website’s data, the neonatal Fc receptor inhibitors and complement inhibitors were effective in treating myasthenia gravis patients in three trials (NCT03315130, NCT03669588, and NCT00727194). This study provides valuable insights into the profile of registered trials on novel therapies for myasthenia gravis. More clinical studies are needed in the future to prove the value of its application.
Similar content being viewed by others

Myasthenia gravis: the changing treatment landscape in the era of molecular therapies
Raffaele Iorio

A path towards personalized medicine for autoinflammatory and related diseases
Jonathan J. Miner & Katherine A. Fitzgerald

Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells
Sangwook Oh, Xuming Mao, … Aimee S. Payne
Introduction
Myasthenia gravis (MG) is an autoimmune disease mediated by antibodies and with the participation of complement, in which antibodies bind to acetylcholine receptors or functionally related molecules in the postsynaptic membrane at the neuromuscular junction 1 . The standard therapies (i.e., cholinesterase inhibitors, corticosteroids, immunosuppressive drugs, immunoglobulin, plasma exchange, and thymectomy) are effective for many MG patients, even though the pathogenic processes of MG are still not fully understood 2 . However, there are still some challenges in treating MG. On the one hand, some refractory patients don’t do well with such traditional therapies 3 . Conversely, some individuals stop using these medicines because of their adverse effects 4 . To address this issue, numerous innovative treatments, particularly those utilizing targeted biological agents (such as neonatal Fc receptor inhibitors, complement inhibitors, and B cell depletors), have emerged and shown promise in recent years 3 . As a result, these drugs have been the subject of an increasing number of clinical trials globally 5 , 6 , 7 . However, to our knowledge, the survey about registered trials on novel therapies for MG is limited. This study aimed to conduct a cross-sectional investigation about this on ClinicalTrial.gov.
Study design and setting
This cross-sectional study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline 8 . The institutional review board’s approval was not required since we conducted this study using publicly available data. Clinicaltrials.gov is the most commonly used clinical trial registration site worldwide, and many studies have used data from this site 9 , 10 . We included trials registered on ClinicalTrials.gov as study subjects.
Definition of novel therapies
Compared with conventional treatments of MG, the novel therapies included targeted biological medications (such as neonatal Fc receptor (FcRn) inhibitor, CD20 B cell depleting agent, complement inhibitor), chimeric antigen receptor T-cell immunotherapy (CAR-T), hematogenic stem cell transplant, etc. This definition served as our inclusion standard (Supplementary Table 1 ).
Data sources and searches
Two investigators (Jinxin Chen and Youtao Wang) independently searched ClinicalTrials.gov. We used words related to MG without any other restrictions. These terms included “myasthenia gravis”, “Myasthenia Gravis, Ocular”, "Ocular Myasthenia Gravis", "Myasthenia Gravis, Generalized", "Generalized Myasthenia Gravis", "Muscle-Specific Receptor Tyrosine Kinase Myasthenia Gravis", "Muscle Specific Receptor Tyrosine Kinase Myasthenia Gravis", "Muscle-Specific Tyrosine Kinase Antibody Positive Myasthenia Gravis", "Muscle Specific Tyrosine Kinase Antibody Positive Myasthenia Gravis", "MuSK myasthenia gravis", "MuSK MG", "Myasthenia Gravis, MuSK", "Anti-MuSK Myasthenia Gravis", "Anti MuSK Myasthenia Gravis" and "Myasthenia Gravis, Anti-MuSK", "Acetylcholine receptor Myasthenia Gravis", "AchR Myasthenia Gravis", "Low-density lipoprotein receptor-related protein 4 Myasthenia Gravis", "LRP4 Myasthenia Gravis", "Agrin Myasthenia Gravis", "Seronegative Myasthenia Gravis", "Bulbar Myasthenia Gravis", "Respiratory Myasthenia Gravis", "Early-onset generalized Myasthenia Gravis", "Late-onset generalized Myasthenia Gravis". All searches were updated until 5th April 2023. The search strategy is as follows: “myasthenia gravis OR Myasthenia Gravis, Ocular OR Ocular Myasthenia Gravis OR Myasthenia Gravis, Generalized OR Generalized Myasthenia Gravis OR Muscle-Specific Receptor Tyrosine Kinase Myasthenia Gravis OR Muscle Specific Receptor Tyrosine Kinase Myasthenia Gravis OR Muscle-Specific Tyrosine Kinase Antibody Positive Myasthenia Gravis OR Muscle Specific Tyrosine Kinase Antibody Positive Myasthenia Gravis OR MuSK MG OR MuSK Myasthenia Gravis OR Myasthenia Gravis, MuSK OR Anti-MuSK Myasthenia Gravis OR Anti MuSK Myasthenia Gravis OR Myasthenia Gravis, Anti-MuSK OR acetylcholine receptor OR AChR OR low-density lipoprotein receptor-related protein 4 OR LRP4 OR Agrin OR seronegative MG OR bulbar MG OR respiratory MG OR early-onset generalized MG OR late-onset generalized MG”.
Trial selection
Supplementary Table 1 lists the inclusion/exclusion criteria. As for inclusion criteria, we included trials using targeted immunotherapies or other biological agents. And we included both interventional and observational trials. For exclusion criteria, we excluded non-myasthenia gravis diseases. Second, we excluded studies using only conventional treatments for MG without novel agents. Thirdly, we ruled out other unrelated treatments. Finally, we eliminated duplicated trials (see Supplementary Table 4 ).
Data extraction
Two reviewers (Jinxin Chen and Youtao Wang) extracted data from the eligible trials independently. Any disagreement regarding the extraction strategy was resolved through discussions. The studied variables included study type, registered year, enrollment, participant age, sponsor, location, center, clinical phenotype, MG autoantibodies, and novel therapies. Also, we gathered information on interventional trials' randomization, blinding, number of arms, assignment, and phase.
Statistical analysis
As this study's primary analysis method, we mainly employed descriptive statistics. Given that the Food and Drug Administration (FDA) authorized the first novel biologic agent for MG in 2017 9 , we compared the characteristics of clinical trials using 2017 as a time boundary. Continuous variables were reported as median (interquartile range, IQR). Categorical data were described as frequency and percentage. The Mann–Whitney and chi-square tests were used to examine differences between clinical trial characteristics before and after 2017. In the summary of clinical trial outcome data, we collected some effect sizes, including mean (standard deviation, SD), least square mean difference (95% confidence interval, CI), mean difference (95% CI), net mean difference (95% CI), and odds ratio (95% CI). R software (version 4.2.1) and Free statistical software (version 1.7.1, FreeClinical Medical Technology Co., Ltd, Beijing, China) were utilized for all analyses. The threshold for statistical significance was a two-sided P value of 0.05.
Ethical standard
The Declaration of Helsinki was followed when conducting the study. We achieved this research utilizing data made available to the public. Therefore, institutional review board permission was not required.
After the initial screening, there were 675 trials on ClinicalTrials.gov in our study. We included 62 studies (registered from 2007 to 2023) for data analysis after discarding 506 trials about non-MG disorders, 41 with only traditional medicines, and 66 unrelated to innovative therapeutics (Fig. 1 ).

Flowchart of trial selection.
To begin with, we conducted a correlation analysis in Fig. 2 between the number of trials and the year that was registered. This result shows that registered trials increase year-on-year (r = 0.76, p < 0.001). Next, we provided a summary of the trial characteristics in Table 1 (for details, see Supplementary Tables 2 and 3 ). Following 2017, more industry-sponsored trials were conducted (91.5% vs. 60%, p = 0.009). Second, there were fewer results on ClinicalTrial.gov after 2017 (10.6% vs. 60%, p < 0.001) (for details, see Supplementary Table 3 6 , 7 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 . Moreover, following 2017, more trials entered phase 3 (67.4% vs. 20%, p = 0.001). Other aspects, including research type, participant age, location, center, publication, blinding method, assignment, and randomization, did not alter after 2017.

Association between the number of trials and registered year. The solid line and the green region represent the predicted value and 95% confidence intervals, respectively. CI confidence interval.
Table 2 illustrates the therapeutic effectiveness of the trials on ClinicalTrial.gov. The findings of three clinical trials indicate some advantages regarding novel therapies for MG. Specifically, in a dose–response control trial (NCT03315130), the RA101495 (Complement inhibitor) group's MG scores dropped more significantly than the placebo group at 12 weeks. For the 0.1 mg/kg group, the least square mean difference (80% CI) for myasthenia gravis activities of daily living (MG-ADL) scale, quantitative myasthenia gravis score (QMGS), 15-item myasthenia gravis quality of life revised scale (MG-QoL15r), and myasthenia gravis composite score (MGCS) were − 2.2 (− 3.9 ~ − 0.5), − 2.3 (− 4.5 ~ − 0.1), − 5.3 (− 8.4 ~ − 2.1) and − 2.0 (− 4.9 ~ 0.9), respectively. Corresponding parts for the 0.3 mg/kg group were − 2.3 (− 4.0 ~ − 0.6), − 2.8 (− 5.1 ~ 0.6), − 3.7 (− 6.9 ~ − 0.6), and − 4.1 (− 7.0 ~ − 1.1). Similarly, another trial (NCT03669588) showed that ARGX-113 (efgartigimod, an FcRn inhibitor) significantly reduced the MG-ADL scale compared to the Placebo, regardless of the AChR-ab status. The Odds Ratio (OR) (95% CI) for the AChR-Ab seropositive individuals and the general population were 4.951 (2.213 ~ 11.528) and 3.699 (1.854 ~ 7.578). And the QMGS dropped more in the ARGX-113 group in anti-AChR MG: the OR was 10.842 (4.179 ~ 31.200). In the other crossover-designed trial (NCT00727194), in both periods, the complement inhibitor eculizumab reduced the MG-ADL scale more than the Placebo: the net mean difference (95% CI) was − 1.58 (− 4.08 ~ 0.91). Additionally, eculizumab dramatically decreased QMGS in period one: the net mean difference (95% CI) was − 4.71 (− 10.80 ~ 1.37) (see more in Supplementary Table 5 ).
Figure 3 compares the number of clinical trials for innovative treatments. The FcRn inhibitor (51.2%) was the most researched medicine on ClinicalTrial.gov. Complement inhibitors (39.0%) and B cell depletors (14.6%) were the second and third, respectively. Other treatments, including IL-6 blockers, CAR T cell therapy, hematopoietic stem cell transplants, cytokines, Bruton's tyrosine kinase (BTK) inhibitors, BAFF inhibitors, anti-CD40 monoclonal antibodies, anti-CD38 monoclonal antibodies haven't been investigated as much.

Distribution of novel therapies. FcRn the neonatal Fc receptor, BAFF B cell-activating factor, CAR-T chimeric antigen receptor T-cell immunotherapy, IL interleukin, BTK Bruton's tyrosine kinase. Other: Orencia (a selective T cell costimulatory immunomodulator); RC18 (TACI-Antibody Fusion Protein), TACI transmembrane activator and calcium-modulator and cyclophilin ligand interactor; CK-2017357 (Tirasemtiv, an activator of the fast skeletal muscle troponin complex); CV-MG01 (Myasterix, a kind of vaccine).
In this study, we outlined the characteristics of the ClinicalTrials.gov-registered trials testing cutting-edge treatments for MG. Additionally, we looked at the treatment effectiveness data from various trials and discovered some significant results. To our knowledge, the related research on this subject is limited. We believe that a thorough examination of the clinical trials of novel MG treatments could significantly change how we approach clinical practice.
According to our research, there has been an increasing number of registered studies of innovative treatments for MG in recent years. It implies that MG treatment is entering a new age marked by precision medicine 34 . Although many MG patients respond well to traditional therapy, these drugs have certain drawbacks 35 . For instance, clinicians frequently face the challenge of how to treat patients with refractory MG due to poor response or severe adverse effects to conventional medications 36 . As a result, many novel therapies have been applied in clinical research 37 .
Also, the characteristics of these trials indicate that the industry is funding an increasing number of trials. Medicine research is more effective with substantial institutional funding and might ease the transfer from basic testing to clinical use 38 . The pharmaceutical industry supports numerous randomized controlled trials, as they are the primary participants in drug discovery and development 11 , 13 . On top of that, our investigation identified more clinical registration trials for MG in Phase 3 following 2017. MG's innovative biologics are progressively being utilized in clinical settings 39 . Despite a rise in Phase 3 clinical trials, their outcomes remain comparatively modest for various reasons. They include difficulties in eligible patient recruitment and poor adherence of some patients to protracted follow-up periods 11 , 18 . Besides, we also found that the percentage of clinical trials with results decreased by 2017. The significant rise in clinical trial registrations could cause this phenomenon. It is well known that any study outcome takes a certain amount of time to complete.
To some extent, it is not surprising that the seemingly contradictory phenomena of fewer results after 2017. Interestingly, no significant difference was found in the relative number of trials published before and after 2017. However, the number of trials published after 2017 (N = 47) was significantly higher than before (N = 15), indicating that there has been a significant increase in the research intensity of novel biologics over the last five years.
The results presented in Table 2 reveal that FcRn and complement inhibitors effectively treat MG. In line with this, Fig. 3 's findings also show that they are the most researched medications on ClinicalTrial.gov. And B cell depletor is another novel biologic that has been extensively studied. Following, we covered some elements of these drugs' mechanism of action and clinical research.
Myasthenia is a type of IgG autoantibody-mediated autoimmune disease. IgG recycling is decreased by inhibiting the FcRn receptor as IgG is degraded in lysosomes 40 . Given that IgG production does not compensate for this decrease, FcRn receptor blockade causes a rapid decline in all IgG subclasses 41 . Efgartigimod is a mutated human IgG1 Fc portion with a strong affinity for binding to FcRn 42 . A phase 3 clinical trial 20 the ADAPT study (NCT03669588) showed that the efgartigimod group had more MG-ADL responders than the placebo group in cycle 1 (2-point improvement in MG-ADL scale lasting for four weeks) (68% vs. 30% p < 0.0001). The OR (95% CI) was 4.95 (2.21 ~ 11.53). Treatment with efgartigimod also resulted in significant and rapid health-related quality-of-life (HRQoL)improvements in generalized MG up to 8 weeks after the first infusion in treatment cycles1(TC1) and TC2 13 . The result shows that FcRn antagonists, represented by efgartigimod, have considerable potential in MG treatment. Notably, efgartigimod has received approval to treat generalized MG globally 43 .
We generally recognize that IgG initiates the complement pathway cascades when it binds to the AChR epitopes. The creation of the C5 convertase marks the culmination of the final steps in this cascade 44 . Eculizumab, a chimeric monoclonal antibody that inhibits the C5 convertase, limits the activation of membrane attack complex (MAC), and reverses the disease status of MG, is one such medication 11 . A global phase 3 clinical trial 11 (REGAIN (NCT 01,997,229)) revealed that eculizumab performed better than the Placebo in terms of the change in QMGS and MG-QOL15 from baseline to week 26 as determined by worst-rank ANCOVA: the differences (95% CI) were − 16.0 (− 28.5 to − 3.4) and − 14.3 (− 27.0 to − 1.6). It means eculizumab offers long-lasting improvements in patients with refractory generalized anti-AChR MG. A tertiary endpoint analysis 19 of the REGAIN open-label extension results found that at week 26 of REGAIN, more eculizumab-treated patients than placebo-treated patients achieved a status of improved (60.7% vs. 41.7%) or minimal manifestations (MM) (25.0% vs 13.3%; standard odds ratio: 2.3; 95% confidence interval: 1.1, 4.5). An analysis 23 which examined changes in the use of immunosuppressive therapy (ISTs) in patients receiving eculizumab during the open-label extension (OLE) of the REGAIN study, found that patients with previously refractory generalized MG used ISTs less frequently (48.7% (57/117)).
Moreover, patients in all groups maintained clinical improvements with eculizumab, including those who decreased or stopped concomitant ISTs. In one subgroup analysis 25 of REGAIN and its OLE study, the researchers conclude that eculizumab treatment results in meaningful clinical improvements and fewer disease exacerbations for patients who previously received chronic IVIg compared with Placebo. In another interim sub-analysis 27 , eculizumab safety in Japanese and Caucasian patients was comparable to the overall REGAIN population. These results show that eculizumab is of great value in the treatment of MG as a representative of complement inhibitors. Furthermore, the FDA approved eculizumab for treating generalized anti-AChR MG in 2017 11 . Zilucoplan and Ravulizumab are undergoing phase 3 investigations, two drugs with mechanisms of action comparable to eculizumab. These studies align with our findings from Table 2 (NCT00727194).
MG is an antibody-mediated disease and depends on B cells to produce pathogenic antibodies 45 . Thus, the focus of medical research on B cells for MG has garnered attention. Rituximab is a CD20 monoclonal antibody that efficiently depletes most B cells, including memory and immature B lymphocytes 46 . According to a systematic review of case reports on 169 individuals, the number of patients with MG relapse after treatment was significantly reduced in both the anti-AChR MG (93% before vs. 26% after) and the anti-Muscle-specific kinase (MuSK) MG (100% vs. 14%) 47 . Beyond rituximab, other medicines targeted specifically at B-cells have been developed. Obinutuzumab provides a distinct mechanism of action from rituximab through primarily direct cell death rather than complement-mediated cytotoxicity. It may be worth considering as an effective treatment for AChR MG 48 .
Furthermore, ofatumumab, ublituximab, and inebilizumab are also anti-B-cell agents with clinical potential in MG 49 . In one observational study, we observed the efficacy and safety of Inebilizumab (an anti-CD19 monoclonal antibody) in treating of MG (NCT04202341). The website does not display this summary. While these drugs, particularly rituximab, have not yet received marketing approval for MG treatment, we believe that as more relevant clinical trials are conducted, they will soon become valuable tools in treating MG.
Researchers are also testing other innovative therapies, including IL-6 blockers 50 , 51 , CAR T cell therapy 52 , and BTK inhibitors 53 . An IL-6 receptor inhibitor, satralizumab, prevents IL-6 signaling, which may impact the pathogenic helper T and B cells in MG 54 . It is the subject of an ongoing global phase 3 clinical investigation (NCT 04,963,270). Similarly, Neutrophils, basophils, monocytes, mast cells, neutrophils, and B cells express BTK. It is essential for B cells' activation, growth, and differentiation 55 . Consequently, BTK inhibitors are becoming prospective treatments for MG and other autoimmune diseases 53 . Since there haven't been many clinical registration trials for these drugs, more clinical study is needed to appreciate their potential fully.
There appears to be a greater prevalence of MG today than before. There could have been some reason for this rise in occurrence. For instance, MG used to increase mortality significantly, but over the years, treatment has improved to the point where life expectancy is now almost average in industrialized nations 56 . Furthermore, the increased use of sensitive tests for MG-specific autoantibodies has improved MG case-finding. A recent study from Japan has revealed a natural rise in incidence, especially for late-onset MG 57 . As a result, the therapeutic demand for MG has increased. It is of great significance that many novel and different agents for MG are going on.
On one hand, traditional ISTs, when used over the long term, bring about specific side effects. However, patients can find relative safety in the new biological agents. On the other hand, a notable percentage, ranging from 10 to 30% of individuals living with MG exhibit varying degrees of resistance to conventional immunosuppression due to the severe side effects from therapy or the presence of persistent and incapacitating weakness 1 . Nonetheless, new agents like rituximab emerge as recommended solutions for refractory MG. Uncontrolled studies revealed that rituximab demonstrated effectiveness across all MG groups, displaying varying response rates 47 , 58 , 59 . Eculizumab, in patients with refractory AChR, also exhibited a noticeable albeit moderately significant efficacy, as demonstrated in the REGAIN study 18 . Furthermore, even though numerous treatment options are now accessible, the challenge confronting physicians is determining the optimal combination of therapies. This selection hinges on predicting efficacy through an assessment of the clinical phenotype and biological markers of the patients.
There are several limitations to our study. Firstly, the study's cross-sectional nature limited us to further causal analysis. Still, we continue to try to learn more about the traits, particularly the effectiveness, of clinical research on new biologics for MG. Besides, our study's representativeness may have a few drawbacks because it only looked at clinical studies registered on the ClinicalTrial.gov website. Nonetheless, we intend to focus our efforts on other online registries.
Here, it is necessary to reemphasize our findings. To begin with, we have found that the growing number of industry-funded clinical trials is beneficial for translating drug development to the clinic. And this encourages more clinician-scientists and research institutions to engage in various forms of collaboration with businesses. Then, although more clinical trials are moving into phase 3, outputs are still only moderately high. The reason includes difficulties with patient recruitment and poor adherence to extended follow-ups. Therefore, it is crucial to address the problem of successfully grounding clinical trial designs. Finally, our investigation found that the most researched novel biologics are FcRn inhibitors, complement inhibitors, and B-cell scavengers. The result indicates that these medications have great promise for both clinical translation and research utility.
What's more, there are some strengths in our study. First, in contrast to other studies, we statistically analyzed the treatment efficacy of the registered trials and came up with some meaningful findings. Second, we used a comprehensive approach by analyzing the collected trials on both a quantitative and qualitative level.
In conclusion, this study might offer helpful information on registered studies of cutting-edge treatments for MG. The findings of this analysis would assist clinical researchers or epidemiologists in conducting more high-quality clinical studies. Future evidence-based medicine will also require more well-designed trials.
Data availability
The publicly available datasets for this work are made available online. Online at https://clinicaltrials.gov/ , you may find the name of the repository or repositories and their accession numbers.
Gilhus, N. E. Myasthenia gravis. N. Engl. J. Med. 375 , 2570–2581 (2016).
Article CAS PubMed Google Scholar
Gilhus, N. E. et al. Myasthenia gravis. Nat. Rev. Dis. Primer 5 , 30 (2019).
Article Google Scholar
Schneider-Gold, C. & Gilhus, N. E. Advances and challenges in the treatment of myasthenia gravis. Ther. Adv. Neurol. Disord. 14 , 175628642110654 (2021).
Verschuuren, J. J. et al. Advances and ongoing research in the treatment of autoimmune neuromuscular junction disorders. Lancet Neurol. 21 , 189–202 (2022).
Nguyen-Cao, T. M., Gelinas, D., Griffin, R. & Mondou, E. Myasthenia gravis: Historical achievements and the “golden age” of clinical trials. J. Neurol. Sci. 406 , 116428 (2019).
Nowak, R. J. et al. Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: The BeatMG study. Neurology 98 , e376–e389 (2022).
Article CAS PubMed PubMed Central Google Scholar
Murai, H. et al. Safety and effectiveness of eculizumab in Japanese patients with generalized myasthenia gravis: interim analysis of post-marketing surveillance. Ther. Adv. Neurol. Disord. 14 , 175628642110019 (2021).
Von Elm, E. et al. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: Guidelines for reporting observational studies. Ann. Intern. Med. 147 , 573 (2007).
Paul, E. et al. Assessing uptake of the core outcome set in randomized controlled trials for Parkinson’s disease: A systematic review. Ageing Res. Rev. 91 , 102081 (2023).
Article PubMed Google Scholar
Terao, I., Honyashiki, M. & Inoue, T. Comparative efficacy of lithium and aducanumab for cognitive decline in patients with mild cognitive impairment or Alzheimer’s disease: A systematic review and network meta-analysis. Ageing Res. Rev. 81 , 101709 (2022).
Howard, J. F. et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 16 , 976–986 (2017).
Allenbach, Y. et al. Efficacy of rituximab in refractory inflammatory myopathies associated with anti-synthetase auto-antibodies: An open-label, phase II trial. PLoS ONE 10 , e0133702 (2015).
Article PubMed PubMed Central Google Scholar
Saccà, F. et al. Efgartigimod improved health-related quality of life in generalized myasthenia gravis: results from a randomized, double-blind, placebo-controlled, phase 3 study (ADAPT). J. Neurol. 270 , 2096–2105 (2023).
Regnault, A. et al. Measuring overall severity of myasthenia gravis (MG): Evidence for the added value of the MG symptoms PRO. Neurol. Ther. https://doi.org/10.1007/s40120-023-00464-x (2023).
Bril, V. et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 22 , 383–394 (2023).
Yan, C. et al. Therapeutic effects of batoclimab in chinese patients with generalized myasthenia gravis: A Double-Blinded, Randomized, placebo-controlled phase II study. Neurol. Ther. 11 , 815–834 (2022).
Piehl, F. et al. Efficacy and safety of rituximab for new-onset generalized myasthenia gravis: The RINOMAX randomized clinical trial. JAMA Neurol. 79 , 1105 (2022).
Siddiqi, Z. A. et al. Eculizumab in refractory generalized myasthenia gravis previously treated with rituximab: subgroup analysis of REGAIN and its extension study. Muscle Nerve 64 , 662–669 (2021).
Mantegazza, R. et al. Post-intervention status in patients with refractory myasthenia gravis treated with eculizumab during REGAIN and its open-label extension. Neurology 96 , e610–e618 (2021).
Howard, J. F. et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): A multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 20 , 526–536 (2021).
Bril, V. et al. Efficacy and safety of rozanolixizumab in moderate-to-severe generalised myasthenia gravis: A phase 2 RCT. Neurology https://doi.org/10.1212/WNL.0000000000011108 (2020).
The REGAIN Study Group et al. ‘Minimal symptom expression’ in patients with acetylcholine receptor antibody-positive refractory generalized myasthenia gravis treated with eculizumab. J. Neurol. 267 , 1991–2001 (2020).
Nowak, R. J. et al. Concomitant immunosuppressive therapy use in eculizumab-treated adults with generalized myasthenia gravis during the REGAIN open-label extension study. Front. Neurol. 11 , 556104 (2020).
Mantegazza, R. et al. Consistent improvement with eculizumab across muscle groups in myasthenia gravis. Ann. Clin. Transl. Neurol. 7 , 1327–1339 (2020).
Jacob, S. et al. Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study. Ther. Adv. Neurol. Disord. 13 , 175628642091178 (2020).
Di Stefano, V. et al. Rituximab in AChR subtype of myasthenia gravis: Systematic review. J. Neurol. Neurosurg. Psychiatry 91 , 392–395 (2020).
Murai, H. et al. Long-term efficacy and safety of eculizumab in Japanese patients with generalized myasthenia gravis: A subgroup analysis of the REGAIN open-label extension study. J. Neurol. Sci. 407 , 116419 (2019).
Muppidi, S. et al. Long‐term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve https://doi.org/10.1002/mus.26447 (2019).
Andersen, H. et al. Eculizumab improves fatigue in refractory generalized myasthenia gravis. Qual. Life Res. 28 , 2247–2254 (2019).
Article ADS PubMed PubMed Central Google Scholar
Yi, J. S., Guptill, J. T., Stathopoulos, P., Nowak, R. J. & O’Connor, K. C. B cells in the pathophysiology of myasthenia gravis: Myasthenia Gravis B cells. Muscle Nerve 57 , 172–184 (2018).
Smith, B. et al. Generation and characterization of a high affinity anti-human FcRn antibody, rozanolixizumab, and the effects of different molecular formats on the reduction of plasma IgG concentration. mAbs https://doi.org/10.1080/19420862.2018.1505464 (2018).
Hewett, K. et al. Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology 90 , e1425–e1434 (2018).
Hehir, M. K. et al. Rituximab as treatment for anti-MuSK myasthenia gravis: Multicenter blinded prospective review. Neurology 89 , 1069–1077 (2017).
Vanoli, F. & Mantegazza, R. Antibody therapies in autoimmune neuromuscular junction disorders: Approach to myasthenic crisis and chronic management. Neurotherapeutics 19 , 897–910 (2022).
Huang, E.J.-C. et al. Myasthenia gravis: Novel findings and perspectives on traditional to regenerative therapeutic interventions. Aging Dis. https://doi.org/10.14336/AD.2022.1215 (2022).
Cortés-Vicente, E. et al. Drug-refractory myasthenia gravis: Clinical characteristics, treatments, and outcome. Ann. Clin. Transl. Neurol. 9 , 122–131 (2022).
Evoli, A. & Damato, V. Conventional and emerging treatments and controversies in myasthenia gravis. Expert Rev. Neurother. 23 , 445–456 (2023).
Parker, G., Hunter, S., Hogarth, S. & Miller, F. A. Industry involvement in evidence production for genomic medicine: A bibliometric and funding analysis of decision impact studies. PLoS ONE 18 , e0285122 (2023).
Su, Y. et al. Knowledge mapping of targeted immunotherapy for myasthenia gravis from 1998 to 2022: A bibliometric analysis. Front. Immunol. 13 , 998217 (2022).
Pyzik, M. et al. The neonatal Fc receptor (FcRn): A misnomer?. Front. Immunol. 10 , 1540 (2019).
Fridman, W. H. Fc receptors and immunoglobulin binding factors 1 . FASEB J. 5 , 2684–2690 (1991).
Gable, K. L. & Guptill, J. T. Antagonism of the neonatal Fc receptor as an emerging treatment for myasthenia gravis. Front. Immunol. 10 , 3052 (2020).
Uzawa, A. & Utsugisawa, K. Biological therapies for myasthenia gravis. Expert Opin. Biol. Ther. 23 , 253–260 (2023).
Defendi, F., Thielens, N. M., Clavarino, G., Cesbron, J.-Y. & Dumestre-Pérard, C. The immunopathology of complement proteins and innate immunity in autoimmune disease. Clin. Rev. Allergy Immunol. 58 , 229–251 (2020).
Rose, N. et al. Receptor clustering and pathogenic complement activation in myasthenia gravis depend on synergy between antibodies with multiple subunit specificities. Acta Neuropathol. (Berl.) 144 , 1005–1025 (2022).
Lee, D. S. W., Rojas, O. L. & Gommerman, J. L. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 20 , 179–199 (2021).
Tandan, R., Hehir, M. K., Waheed, W. & Howard, D. B. Rituximab treatment of myasthenia gravis: A systematic review: Rituximab in Myasthenia Gravis. Muscle Nerve 56 , 185–196 (2017).
Ingelfinger, F. et al. Antibodies produced by CLL phenotype B cells in patients with myasthenia gravis are not directed against neuromuscular endplates. Neurol. Neuroimmunol. Neuroinflamm. 10 , e200087 (2023).
Greenfield, A. L. & Hauser, S. L. B-cell therapy for multiple sclerosis: Entering an era: MS: Entering the era of B-cell therapy. Ann. Neurol. 83 , 13–26 (2018).
Uzawa, A. et al. High levels of serum interleukin-6 are associated with disease activity in myasthenia gravis. J. Neuroimmunol. 358 , 577634 (2021).
Jonsson, D. I., Pirskanen, R. & Piehl, F. Beneficial effect of tocilizumab in myasthenia gravis refractory to rituximab. Neuromuscul. Disord. 27 , 565–568 (2017).
Su, M., Zhao, C. & Luo, S. Therapeutic potential of chimeric antigen receptor based therapies in autoimmune diseases. Autoimmun. Rev. 21 , 102931 (2022).
Esfandiari, E. et al. A phase I, randomized, double-blind, placebo-controlled, single-dose and multiple-rising-dose study of the BTK inhibitor TAK-020 in healthy subjects. Clin. Transl. Sci. 14 , 820–828 (2021).
Menon, D. & Bril, V. Pharmacotherapy of generalized myasthenia gravis with special emphasis on newer biologicals. Drugs 82 , 865–887 (2022).
Ringheim, G. E., Wampole, M. & Oberoi, K. Bruton’s tyrosine kinase (BTK) inhibitors and autoimmune diseases: Making sense of BTK inhibitor specificity profiles and recent clinical trial successes and failures. Front. Immunol. 12 , 662223 (2021).
Gilhus, N. E. & Verschuuren, J. J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 14 , 1023–1036 (2015).
Zieda, A. et al. A nationwide epidemiological study of myasthenia gravis in Latvia. Eur. J. Neurol. 25 , 519–526 (2018).
Robeson, K. R. et al. Durability of the rituximab response in acetylcholine receptor autoantibody-positive myasthenia gravis. JAMA Neurol. 74 , 60 (2017).
Anderson, D., Phan, C., Johnston, W. S. & Siddiqi, Z. A. Rituximab in refractory myasthenia gravis: A prospective, open-label study with long-term follow-up. Ann. Clin. Transl. Neurol. 3 , 552–555 (2016).
Download references
Acknowledgements
We greatly appreciate Qilin Yang from the Department of Critical Care, the Second Affiliated Hospital of Guangzhou Medical University, Guangdong, China, with the comments regarding the manuscript.
Author information
These authors contributed equally: Xingyue Li and Jinxin Chen.
Authors and Affiliations
Department of Neurology, Xiangyang No.1 People’s Hospital, Hubei University of Medicine, Xiangyang, China
Hubei University of Medicine, Shiyan, China
Jinxin Chen, Youtao Wang, Siwei Zheng & Kun Wan
Department of Neurology, Taihe Hospital, Hubei University of Medicine, Shiyan, China
Xiaodong Liu
You can also search for this author in PubMed Google Scholar
Contributions
X.L.: Writing—original draft; Writing—review & editing. J.C.: Data collection; Formal analysis; Writing—review & editing. Y.W.: Data collection; Writing—review & editing. S.Z.: Data collection; Writing—review & editing. K.W.: Data collection; Writing—review & editing. X.L.: Conceptualization; Methodology; Supervision; Writing—review & editing.
Corresponding author
Correspondence to Xiaodong Liu .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary table 1., supplementary table 2., supplementary table 3., supplementary table 4., supplementary table 5., rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Li, X., Chen, J., Wang, Y. et al. Registered trials on novel therapies for myasthenia gravis: a cross-sectional study on ClinicalTrials.gov. Sci Rep 14 , 2067 (2024). https://doi.org/10.1038/s41598-024-52539-w
Download citation
Received : 10 July 2023
Accepted : 19 January 2024
Published : 24 January 2024
DOI : https://doi.org/10.1038/s41598-024-52539-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
By submitting a comment you agree to abide by our Terms and Community Guidelines . If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

REVIEW article
The intricate dance of non-coding rnas in myasthenia gravis pathogenesis and treatment.

- Department of Neurology, The First Affiliated Hospital of China Medical University, Shenyang, China
Myasthenia gravis (MG) stands as a perplexing autoimmune disorder affecting the neuromuscular junction, driven by a multitude of antibodies targeting postsynaptic elements. However, the mystery of MG pathogenesis has yet to be completely uncovered, and its heterogeneity also challenges diagnosis and treatment. Growing evidence shows the differential expression of non-coding RNAs (ncRNAs) in MG has played an essential role in the development of MG in recent years. Remarkably, these aberrantly expressed ncRNAs exhibit distinct profiles within diverse clinical subgroups and among patients harboring various antibody types. Furthermore, they have been implicated in orchestrating the production of inflammatory cytokines, perturbing the equilibrium of T helper 1 cells (Th1), T helper 17 cells (Th17), and regulatory T cells (Tregs), and inciting B cells to generate antibodies. Studies have elucidated that certain ncRNAs mirror the clinical severity of MG, while others may hold therapeutic significance, showcasing a propensity to return to normal levels following appropriate treatments or potentially foretelling the responsiveness to immunosuppressive therapies. Notably, the intricate interplay among these ncRNAs does not follow a linear trajectory but rather assembles into a complex network, with competing endogenous RNA (ceRNA) emerging as a prominent hub in some cases. This comprehensive review consolidates the landscape of dysregulated ncRNAs in MG, briefly delineating their pivotal role in MG pathogenesis. Furthermore, it explores their promise as prospective biomarkers, aiding in the elucidation of disease subtypes, assessment of disease severity, monitoring therapeutic responses, and as novel therapeutic targets.
1 Introduction
Myasthenia gravis (MG) is an autoimmune disorder affecting the neuromuscular junction, primarily instigated by the presence of antibodies targeting various postsynaptic components ( 1 ). Among these antibodies, anti-acetylcholine receptor antibodies (AChR-Ab) stand as the most prevalent, while antibodies against MuSK (MuSK-Ab), Lrp4, and agrin are comparatively less common ( 2 ). Additionally, biomarkers such as ColQ, Kv1.4, titin, and RyR are indeed valuable, albeit their specific pathogenic roles remain uncertain ( 3 ). The hallmark feature of MG is fatigable muscle weakness, accentuated post-exertion and relieved during rest ( 4 ). MG classification encompasses subgroups based on antibody profiles, clinical manifestations, age of onset, and thymus pathology ( 2 ). Notably, thymus involvement plays a pivotal role in AChR-Ab-positive MG (AChR-MG), with hyperplasia observed in early-onset MG (EOMG) and atrophy in late-onset MG (LOMG) ( 2 ). Thymoma-associated MG (TAMG) represents a distinct subgroup ( 2 ). The advent of high-throughput sequencing technology has redefined our perception of ncRNAs, once considered “junk DNA,” as critical cellular regulators ( 5 ). This vast class of genomic elements is typically categorized into two main groups: small or short non-coding RNAs and long non-coding RNAs (lncRNAs), based on whether their length exceeds 200 nucleotides ( 6 ). Additionally, circular RNAs (circRNAs), unique single-stranded RNA molecules formed by covalent closure at the 5’ and 3’ ends, have emerged as noteworthy constituents of ncRNAs ( 7 ). Intriguingly, ncRNAs have exhibited profound associations with diverse diseases, spanning cancer, cardiovascular and cerebrovascular conditions, as well as metabolic and autoimmune disorders ( 8 – 10 ). In systemic lupus erythematosus (SLE), some miRNAs, such as miR-125a, miR-125b, miR-21, miR-148a, miR,223, and miR-31, expressed abnormally, others, like miR-7, miR-155, miR-146, and miR-182 were reported to regulate B cells, T cells, or the formation of germinal centers (GCs) ( 11 – 13 ). While in rheumatoid arthritis (RA), miR-146a, miR-155, miR-22, and miR-10a-5p were related to the inflammatory environment ( 13 ). Let-7g-5p and NEAT1, a lncRNA, were proved to promote the proliferation of Th17 ( 14 ). With this backdrop, the investigation of ncRNAs on MG pathogenesis has become a compelling area of study. Despite significant advancements in MG management, several challenges persist ( 2 ). The etiological “triggers,” underlying molecular mechanisms, and regulatory factors at the molecular level remain elusive, confounding researchers ( 15 ). Furthermore, a subset of MG patients fails to achieve remission or substantial clinical improvement with current immunotherapies ( 16 ), a concern exacerbated by the drawbacks of long-term immunosuppressant usage, including intolerance, delayed therapeutic onset, and systemic toxicity ( 17 ). Our conviction lies in the potential of precision-targeted immunotherapy as a promising avenue to tackle these challenges. In this review, we delve into the role of ncRNAs in MG pathogenesis and explore the significance of ncRNAs in MG treatment and therapeutic monitoring.
2 miRNAs in MG
Among the diverse categories of small non-coding RNAs, microRNAs (miRNAs) supreme as one of the most prominent classes, distinguished by their sheer number and prevalence in research ( 18 ). These endogenous single-stranded molecules typically comprise approximately 22 nucleotides ( 19 ) and were initially discovered by Lee et al. in the early 1990s ( 20 ). In the realm of immune system modulation, miRNAs play an integral role, which explains why disruptions in their function have been associated with a range of autoimmune conditions, such as MG ( 21 , 22 ) ( Table 1 ).

Table 1 Differential expressed miRNAs in MG.
2.1 miRNAs involved in MG pathogenesis
In patients with MG, a series of dysfunctions occur at the neuromuscular junction (NMJ), driven by T cells and mediated by B cells, resulting in a complex pathogenic process ( 24 ). This process involves an array of miRNAs and proinflammatory cytokines, potentially contributing to the development of MG ( Figure 1 ).

Figure 1 Dysregulated ncRNAs in MG pathogenesis. Abnormally expressed ncRNAs in different types of samples actively participate in the pathogenesis of AChR-MG by regulating the differentiation of T cells, broking the balance between helper T cells (Th1 and Th17) and Treg cells, activating the B cells, modulating the production of inflammatory cytokines, thus promoting the production of antibodies. Here, we delineate the regulatory relationship between abnormally expressed ncRNAs and immune cells, cytokines, and related signaling pathways.
Several studies have explored the role of dysregulated miRNAs in MG by compiling a catalog of genes associated with MG susceptibility based on existing literature ( 27 , 28 , 53 , 56 ). Yang et al. ( 28 ) conducted an analysis of 93 miRNAs associated with MG risk pathways, shedding light on their regulatory role within these pathways. Their work underscored the potential significance of some specific miRNAs in MG pathogenesis, including miR-497, miR-15a, miR-15b, miR-16, and miR-195. Cao et al. ( 56 ) pinpointed miRNA-146a as an important contributor to the regulation of numerous MG risk pathways, underscoring its pivotal role in MG pathogenesis. Moreover, in the study of Bo et al. ( 53 ), 13 dysregulated miRNAs, including miR-29b-3p, miR-145-5p, and miR-451a, were believed to play a role in MG pathogenesis. Among these risk-related miRNAs, miR-145-5p was the sole miRNA found to exhibit differential expression, suggesting its potential pivotal function in MG. In a complementary study, Qian et al. ( 27 ) reported 30 abnormally expressed miRNAs, which may participate in MG development by modulating Tregs. Collectively, these identified miRNAs offer valuable insights and may serve as guiding principles for future research endeavors aimed at unraveling the intricate pathogenesis of MG.
2.1.1 Modulation of Tregs, Th17 and Th1
It is well-established that the initiation of MG critically hinges on the activation of autoreactive T cells ( 57 ). Consequently, the miRNAs exhibiting abnormal expression and responsibility for T cell proliferation and activation may represent a pivotal link in MG pathogenesis. Certain miRNAs have been identified to be involved in the regulation of T cells in MG. Cron et al. ( 58 ) discovered that miR-150 could target MYB, a proto-oncogene, thus affecting the survival of CD4 + and CD8 + T cells, which may contribute to the maintenance of an immunologically activated state. Research showed that miR-20b would inhibit T cell proliferation and activation by suppressing the NFAT signaling pathway through downregulating NFAT5 and CAMTA1 ( 33 ). Moreover, decreased miR-522-3p can lead to the overexpression of CD25, CD69, IL-2, and IL-10 by targeting SLC31A1, consequently promoting the activation and proliferation of Jurkat cells, a T cell leukemia line ( 31 ). Dysregulation of key immune cell populations, including Tregs, Th17, and Th1, exerts a significant influence on MG pathogenesis. Studies have reported that the expression of Tregs is associated with disease severity, with observations of decreased Foxp3, a Treg-related cytokine, in peripheral blood mononuclear cells (PBMCs) of MG patients ( 39 ). In an AIRE knockout mouse model, reduced Tregs and increased Th17 cells in the thymus rendered the mice more susceptible to experimental autoimmune myasthenia gravis (EAMG) ( 42 ), mirroring the pathogenesis of LOMG and TAMG ( 15 ). Villegas et al. ( 46 ) noted that an imbalance between Tregs and Th17 cells is associated with chronic thymic inflammatory conditions in AChR-MG.
The lapses in Treg function result in the loss of their inhibitory effect on excessive inflammatory responses, leading to uncontrolled inflammatory cell activity in MG ( 25 ). This unchecked immune activity gives rise to chronic inflammation and thymic hyperplasia, further disrupting immune tolerance mechanisms. Recent research outcomes have unveiled a miR-146b-TRAF6-NF-κB-Foxp3 pathway, which plays a crucial role in suppressing Treg proliferation and its inhibitory function ( 49 , 59 ). Intriguingly, Yan et al. ( 60 ) proposed that overexpressed miR-146a may contribute to the pathogenesis of MG by promoting TRAF6 expression, which could further disrupt Treg function. miR-125a-5p was confirmed to negatively regulate Foxp3, consequently inhibiting the generation of Treg with a great possibility ( 37 ). Additionally, there is a positive correlation between the reduced miR-126 and Foxp3 mRNA in MG, providing strong evidence for the downregulation of Treg activity ( 50 ). In summary, differentially expressed miRNAs in MG have been observed to inhibit Treg proliferation and weaken their inhibitory function. Consequently, these miRNA-mediated changes impair immunological tolerance mechanisms and play an important role in the pathogenesis of MG.
Compelling scholarly evidence has demonstrated that the activation of Th17 and Th1 cells, when losing the control of Tregs, can lead to the release of pro-inflammatory cytokines such as INF-γ and IL-17 ( 61 ), which may build the bridge between some dysregulated miRNAs and MG. For instance, Wang et al. ( 62 ) revealed that miR-145 could serve as a regulator that promotes T cell proliferation and differentiation into Th17 cells, by targeting CD28 and NFATc1. Similarly, Cron et al. ( 34 ) reported a reduction of the miR-29 family within thymic tissues of MG, potentially in connection with an increase of IFN-β. Among these, miR-29a/b1, part of a miR-29a genomic cluster, could possibly facilitate the flourishing of Th17 cells. Besides, miR-19b-5p in TAMG has been shown to post-transcriptionally inhibit thymic stromal lymphopoietin (TSLP), thereby regulating Th17 cells and their related cytokines ( 44 ). It’s reasonable to speculate that these abnormally expressed miRNAs may function as regulators, promoting the release of Th17-related cytokines (IL-6, TGF-β, IL-17, IL-1β, and IL-23) and the proliferation of Th17 cells. On the grounds of research by Cheng et al. ( 45 ), with the target of MAPK1, miR-320a may influence the Th1-associated cytokines, like IL-2, and IFN-γ, by adjusting COX-2 through the regulation of ERK/NF-κB pathways. In addition, several miRNAs contribute to the pathogenesis of MG by influencing both Th1 and Th17 responses concurrently. Liu et al. ( 63 ) reported that miR-15a could affect the CXCL10 gene by regulating Th1- and Th17-related cytokines to generate immune responses. In conclusion, the dysregulation of miRNAs can disrupt Treg function, promote the proliferation of Th1 and Th17 cells, and result in the overexpression of pro-inflammatory cytokines, thus modulating the pathogenesis of MG.
2.1.2 miRNAs as regulators of B cell activation
B cells, integral players in the pathogenesis of MG, participate in the formation of ectopic GCs, receive signals from antigen-presenting cells (APCs), and contribute to the production of autoimmune antibodies ( 38 ). Many dysregulated miRNAs can modulate chemokines and specific signaling pathways to promote the maturation of autoreactive B cells and the development of GCs.
miR-155, found to be elevated in B cells, engaged in the immune response through the BAFF-R/TRAF3/NIK/NF-κB p65 pathway, mediating the survival of activated B cells and increasing the production of AChR antibodies by regulating co-stimulatory molecules ( 48 ). The binding of BAFF to BAFF-R is necessary for B cell maturation and survival ( 43 ). And miR-155 can promote the expression of BAFF-R and TRAF3 to facilitate the phosphorylation of NIK, thus helping the NF-κB to translocate into the nuclear of B cells ( 48 ). At the same time, miR-155 can also modulate the co-stimulatory molecules, such as CD40, CD80, and CD86, to enhance the production of antibodies by B cells. Two independent studies mentioned an elevation of miR-146a in AchR-specific B cells and they also reported that miR-146a could affect B cell immunity ( 64 , 65 ). Notably, one of these studies proposed that miR-146a may facilitate the activation of B cells and the production of antibodies by modulating the TLR4 and NF-κB pathways ( 64 ). In a study by Bortone et al. ( 66 ), the downregulated miR-146a showed a noteworthy negative correlation with IRAK1, c-REL, ICOS, and FAS in the follicular hyperplastic thymus of EOMG. The genes targeted by miR-146a appear to collectively contribute to the activation of B cells and the formation of GCs. Firstly, the downregulation of miR-146a allows IRAK1 to induce excessive inflammation and disrupt immune tolerance through the TLR signaling pathway. Secondly, the deficiency of miR-146a can enhance the activation of c-REL, thereby promoting the proliferation and differentiation of B cells and amplifying GCs formation. Furthermore, reduced miR-146a may fail to effectively restrict the aggregation of follicular helper T cells (Tfhs) and the proliferation of B cells in GCs by targeting ICOS. Lastly, miR-146a can downregulate FAS expression, promoting lymphoproliferation and GCs formation. Wang et al. ( 67 ) found that miR-181a may influence the levels of inflammatory cytokines, such as TNF-α, IL-4, and IL-6, affecting B cell proliferation by regulating TRIM9.
Additionally, miR-548k, reduced in the thymus of MG, can target CXCL13 ( 40 ), which plays a role in directing B cells and facilitating GCs formation. miR-452-5p and miR-139-5p were associated with the promotion of RGS13 expression, leading to B cell proliferation and GCs expansion ( 54 ). miR-150-5p has shown a positive correlation with CD19 + and CD27 + B cells, suggesting its involvement in B cell differentiation, memory B cell formation, and further possible immune responses, including B cell activation and antibody production ( 30 ). Additionally, the abnormally expressed miR-126 and miR-21 in PBMCs were proved to upregulate the expression of IL-6, thus promoting the proliferation and differentiation of B cells, consequently facilitating the production of antibodies ( 50 ). let-7c was reported to target IL-10, probably participating in MG pathogenesis by stimulating B cells ( 68 ). Besides, the reduced miR-7 in thymus was reported to upregulate the expression of CCL21, thus helping the formation of GCs ( 69 ).
2.2 miRNAs as clinical biomarkers and therapeutic targets
2.2.1 mirnas engaged in achr-mg.
AChR-Ab is the most prevalent antibody in MG, accounting for approximately 85% of cases ( 70 ). AChR-MG can be further classified according to clinical features. Interestingly, there are some differences in thymus pathology among EOMG, LOMG and TAMG. EOMG refers to individuals under the age of 50, characterized by hyperplastic thymus pathology. In contrast, LOMG manifests in individuals over the age of 50, commonly associated with thymic atrophy. TAMG, on the other hand, represents a paraneoplastic syndrome of thymoma ( 4 ). A few of studies ( 54 , 55 , 58 , 64 , 66 , 69 , 71 ) revealed some differentially expressed miRNAs, such as miR-139-5p, miR-452-5p, miR-612, miR-3654, miR-365, miR-150, miR-20b, miR-192, miR-7, miR-125a-5p, and miR-146a, in the AChR antibody-positive EOMG (AChR-EOMG) patients. A decrease in miR-15b expression was observed in the serum of both early-onset and late-onset AChR-MG ( 71 ), which aligns with findings from an animal model of EAMG in mice injected with Torpedo AChR ( 72 ). In addition, in patients with AChR antibody-positive LOMG (AChR-LOMG), miR-122, miR-140-3p, miR-185, miR-885-5p, miR-106b-3p, miR-223-5p, miR-140-5p, miR-19b-3p, miR-30e-5p, and miR-150-5p were found to express abnormally ( 29 , 32 , 71 ). These findings suggest a nuanced expression profile of dysregulated miRNAs between EOMG and LOMG. Many miRNAs were reported to express aberrantly in TAMG, including miR-125a-5p, miR-19b-5p, miR-20b and miR-522-3p ( 25 , 33 , 50 , 58 ). Additionally, a study by Shi et al. ( 72 ) indicated a decrease in miR-15b expression in TAMG, EOMG, and LOMG patients.
Several dysregulated miRNAs in AChR-MG, compared with ocular myasthenia gravis (OMG), have more severe abnormalities in generalized myasthenia gravis (GMG), some of which are correlated with quantitative myasthenia gravis scores (QMGs) or myasthenia gravis composite scores (MGCs), reflecting the severity of MG and further serving as potential biomarkers for disease surveillance with great possibility. Research findings indicated that both OMG and GMG patients showed elevated miR-150-5p, miR-21-5p, and miR-30e-5p, which had a positive correlation with MGCs and significantly more overexpressed in GMG than in OMG ( 29 , 32 ). Conversely, the lower expressed miR-181a, miR-106a-5p, and miR-20b in MG, had a negative relationship between QMGs, with GMG displaying notably reduced levels compared to OMG ( 73 – 75 ). Additionally, several studies ( 51 , 76 ) on curative effect indicated a parallel between the reduction of AChR-Ab titers and the improvement of symptoms. A fast decrease of AChR-Ab titers more than 50% also made a clinical sense ( 23 ). Interestingly, some studies showed that miR-126, and miR-145 were negatively correlated with AChR-Ab titer ( 50 , 62 ), while miR-21 was positively related to AChR-Ab titer ( 50 ). We therefore speculate that specific ncRNAs may also be useful for efficacy detection.
MiR-150-5p, miR-30e-5p, and miR-146a levels in the serum of AChR-MG patients have shown promising responses to various treatments like thymectomy and immunosuppression, indicating a correlation between their levels and clinical improvement ( 26 , 30 , 32 , 35 , 52 , 58 , 66 ). These miRNAs could potentially serve as biomarkers for monitoring therapeutic efficacy and reflecting the disease severity. Furthermore, research by Zhang et al. ( 65 ) suggested that silencing miR-146a could reduce the expression of CD40, CD80, and CD86 on the surface of B cells, inhibit B cell differentiation, and consequently reduce the production of anti-AChR antibodies. By comparing the level of miR-146a and the expression of c-REL in the thymus of MG in corticosteroid-naïve patients and corticosteroid-treated patients, Bortone et al. ( 66 ) reported that after corticosteroid treatment, the immune response of c-REL, which was originally active in GCs and infiltrating B cells of untreated patients, was strongly inhibited, highlighting the miR-146a/c-REL axis as a potential therapeutic target for immunosuppressants. These discoveries suggests that miR-146a could serve as both a therapeutic target and a monitoring indicator for treatment efficacy. Moreover, Wang et al. ( 48 ) demonstrated that silencing miR-155 could inhibit the production of anti-T-AChR antibodies, making miR-155 another potential treatment target. Sengupta et al. ( 54 ) proposed that miR-139-5p and miR-452-5p might be considered for MG treatment, especially for EOMG, as their mimics inhibit B cell chemotaxis and GCs formation. Additionally, Cavalcante et al. reported that AChR-MG with abnormally expressed miRNAs such as miR-323b-3p, miR-409-3p, miR-485-3p, miR-181d-5p, and miR-340-3p are not sensitive to immunosuppressants ( 47 ), suggesting the presence of these miRNAs may result in poor treatment. In other words, these miRNAs may be used to forecast clinical response.
In essence, miRNAs exhibit subtle variations in expression across MG subgroups. Certain miRNAs with abnormal expression levels have the potential to facilitate disease monitoring, aid in targeted therapy, and assess treatment efficacy. Firstly, several miRNAs tend to normalize after appropriate treatment. Secondly, certain miRNAs may be implicated in antibody production, making them potential targets for MG treatment. Lastly, the presence of specific miRNAs may indicate the response to treatment. These miRNAs hold promise as specific biomarkers reflecting disease severity or treatment responses and may serve as novel targets for MG therapy. Consequently, there arises the prospect of precision therapy and personalized monitoring for individuals with MG.
2.2.2 miRNAs intertwined with MuSK-MG
MuSK antibodies are present in approximately 1-10% of MG cases, predominantly affecting young women under the age of 40 ( 77 ). Although research on MuSK-Ab-positive MG (MuSK-MG) has not been as extensive as that on AChR-MG, several studies have reported differentially expressed miRNAs in this subgroup. Sabre et al. ( 36 ) observed a significant decrease in miR-210-3p and miR-324-3p levels in the serum of MuSK-MG. Punga et al. ( 78 ) identified differential expression of let-7a-5p, let-7f-5p, miR-151a-3p, and miR-423-5p in the serum of MuSK-MG compared to healthy controls. An investigation of PBMCs in MuSK-MG revealed 5 overexpressed and 96 under expressed miRNAs, with marked decreases observed in miR-340-5p, miR-106b-5p, miR-27a-3p, and miR-15a-3p ( 79 ). Moreover, an animal study identified 13 abnormally expressed miRNAs in the omohyoid muscle of MuSK-Ab-positive EAMG mice, particularly highlighting the elevation of miR-1933-3p and miR-1930-5p ( 80 ). Additionally, in a therapeutic study, the serum level of miR-151a-3p in MuSK-MG patients decreased after treatment ( 81 ), indicating a possibility for miR-151a-3p as potential therapeutic target and monitor for treatment efficacy in MuSK-MG.
Unlike AChR and MuSK antibodies, Lrp4 and agrin antibodies are rare in MG, resulting in limited research on these subgroups. Although some miRNAs have been identified as potential regulators of Lrp4 or agrin ( 82 – 88 ), the precise relationship between miRNAs and these two antibodies remains undiscovered ( 89 ).
3 lncRNAs in MG
Being different from miRNAs, lncRNA genes exhibit low conservation across evolution ( 90 ). They play a pivotal role in modulating the immune system through various mechanisms, such as guiding the development of immune cell lineages, orchestrating dynamic transcriptional programs that activate immune cells ( 91 ), and regulating immune-related genes ( 92 ). As an autoimmune disease, MG has a close connection with lncRNAs ( Table 2 ).

Table 2 Differential expressed lncRNAs in MG.
3.1 The role of lncRNAs in MG pathogenesis
LncRNAs have the capacity to modulate the balance between T cell subtypes and regulate proinflammatory cytokines, thereby participating in the pathogenesis of MG ( Figure 1 ). Interestingly, some lncRNAs can serve as the ceRNA, certain RNA sequestering another RNA to influence its primary targets through microRNA response elements (MREs) ( 95 ) to regulate the development of MG.
Several studies have aimed to construct lncRNA-related networks to uncover potential relationships among genes, ncRNAs, and signaling pathways, thus shedding light on MG pathogenesis. For instance, Xu et al. ( 104 ) identified LINC00173, FAM13A-AS1, and OIP5-AS1 as closely associated with phosphatase and tensin homolog (PTEN), with LINC00173 showing promise as a potential MG biomarker. Another study by Lu et al. ( 97 ) suggested that lncRNAs such as NR_104677.1, NR_022008.1, and ENST00000581362.1 may play a role in MG progression by acting as miRNA sponges in specific tuples involving miRNAs like miR-15b-5p and miR-146a-5p. Hong et al. ( 93 ), in their research involving culturing human primary myoblast cells with AChR antibodies, established a co-expressed network of lncRNAs and protein-coding RNAs. They found that MEG3, RP11-184M15.1, and SNHG3 were co-expressed with several protein-coding RNAs, and MEG3, in particular, was linked to cellular homeostasis pathways. The dysregulation of MEG3 might contribute to the development of MG by disrupting cellular equilibrium.
3.1.1 Regulation of immune cells and cytokines
Just like miRNAs, dysregulated lncRNAs can also influence pro-inflammatory cytokines and associated signaling pathways, promoting the proliferation of CD4+ T cells and B cells, inhibiting the proliferation of Treg cells, and activating Th17 and Th1 cells to disrupt immune homeostasis in MG.
In the study by Xu et al. ( 98 ), GAS5 was found to decrease in CD4 + T cells and directly negatively regulate the expression of miR-23a. Furthermore, overexpressed GAS5 was shown to disrupt the balance between Th17 and Treg cells, restraining Th17 differentiation by sponging miR-23a. Another related study ( 103 ) indicated that the upregulation of GAS5 was associated with increased levels of IL-10, coinciding with an improvement in MG symptoms. Two studies by Hu et al. ( 94 , 102 ) demonstrated that XLOC_003810 was elevated in thymic CD4 + T cells in MG. In one study, overexpression of XLOC_003810 in TAMG disrupted the balance between Treg and Th17 by favoring Th17 differentiation and increasing Th17-associated markers such as RORγt, IL-6, and IL-17, while reducing Treg-related markers like Foxp3, TGF-β1, and IL-10. Another study emphasized that XLOC_003810 promoted the expression of CD4 + T cells and their inflammatory cytokines, such as IFN-γ, TNF-α, and IL-1β, highlighting its significant role in MG pathogenesis, especially in TAMG. Luo et al. ( 100 ) found lower levels of IFNG-AS1 in PBMCs of MG, which were negatively correlated with HLA-DOB and HLA-DRB1. They also demonstrated that increased IFNG-AS1 could suppress the proliferation of Th1 cells and promote the expansion of Treg cells, along with some of their transcription factors. IFNG-AS1 was considered to downregulate CD40L and T-bet in CD4 + T cells of MG, partly dependent on HLA-DRB1, implying the involvement of IFNG-AS1 in CD4 + T-related immune responses. HCG18, an upregulated ceRNA in PBMCs, sponging miR-145-5p to modulate CD28, was proved to inhibit apoptosis and enhance the proliferation of Jurkat cells ( 99 ). Moreover, OIP5-AS1 was also verified to have a similar effect on Jurkat cells in another study ( 105 ), which can be achieved through regulating IL-7 by sponging miR-181c-5p. Additionally, Wang et al. ( 101 ) demonstrated that the upregulated SNHG16 can serve as a ceRNA, competitively binding with let-7c-5p in PBMCs of MG. This action not only facilitates Jurkat cell proliferation and inhibits their apoptosis but also influences the level of IL-10, potentially further promoting the activation of B cells.
3.2 Clinical prospects and therapeutic potential of lncRNAs
In AChR-MG, some of the abnormally expressed lncRNAs are closely related to QMGs and MG Impairment Index (MGII), making them indicative of disease severity, while others may aid in subgroup diagnosis, particularly for TAMG. Luo et al. ( 100 ) found that IFNG-AS1 in PBMCs exhibited significant negative correlations with QMGs. Besides, GAS5 was also observed to have strong associations with QMGs and MGII ( 103 ). In another expression profile, five lncRNAs, NR_104677.1, ENST00000583253.1, NR_046098.1, NR_022008.1, and ENST00000581362.1, were reported to remarkedly overexpress in MG exosome, wherein, NR_046098.1 was upregulated prominently with the severity of MG ( 97 ). Another study profiling lncRNA expression identified numerous dysregulated lncRNAs in PBMCs ( 96 ). Among them, ENSG00000250850.2, ATP6VOE2-AS1, and XLOC_000734 were the three most significantly downregulated lncRNAs, while XLOC_003810, XLOC_005780, and ENSG00000259354.1 were the top three highly upregulated lncRNAs when compared to healthy controls. In the context of TAMG, there were 3,699 upregulated lncRNAs and 661 downregulated lncRNAs identified. Notably, XLOC_006297 exhibited the highest expression, while ENSG000000218510.3 had the lowest expression levels among the identified lncRNAs ( 106 ). In a study involving 34 MG patients and 13 healthy controls, Luo et al. ( 107 ) identified significant dysregulation of lncRNAs in PBMCs. Their findings revealed distinct expression patterns when comparing different experimental and control groups. Some of their key observations relied on the fact that, compared with the control group, oebiotech_11933 and A_24_P927716 were the top two overexpressed lncRNAs, while A_21_P0010030 and A_21_P0002844 were the least expressed in TAMG patients. They have also verified that the expression of A_19_P00315959 and oebiotech_13222 in TAMG was much more elevated than that in non-thymoma MG, while the level of oebiotech_22652 and oebiotech_16223 in TAMG patients was much lower. Finally, oebiotech_11933 and oebiotech_03926 were significantly upregulated, whereas ebiotech_02627 and oebiotech_22482 were substantially reduced in MG patients without thymoma versus healthy controls.
In addition to serving as biomarkers, the dysregulated lncRNAs also have the potential to be targeted for treatment. Kong et al. ( 108 ) suggested that MALAT-1 may function as an endogenous sponge, competing with male-specific lethal 2 (MSL2) to bind miR-338-3p. This interaction could lead to the inhibition of T cells and potentially play a protective role, making the MALAT-1-miR-338-3p-MSL2 network a promising therapeutic target.
4 circRNAs in MG
circRNAs were first reported in viroids by Sanger et al. ( 109 ) in 1976. These molecules, characterized by their circular structure, can be categorized into four main types ( 110 ): exonic circRNAs (ecircRNAs), exon-intron circRNAs (EIciRNAs), intronic circRNAs (ciRNAs), and tRNA intronic circular RNAs (tricRNAs). While the functions of most circRNAs remain poorly understood, they are known to play crucial roles in immune regulation, including the adjustment of immune cells, handling immune responses, and modulating immune signaling pathways ( 111 ).
Recent research has begun to shed light on aberrant circRNAs in MG ( Table 3 ), although only a limited number of studies have explored the connection between circRNAs and MG to date. These abnormally expressed circRNAs may contribute to the pathogenesis of MG, reflect disease severity, and present a potential as therapeutic targets.

Table 3 Differential expressed circRNAs in MG.
Two published studies on circRNAs and MG have identified several circRNAs with altered expression patterns that may participate in MG pathogenesis and serve as potential biomarkers: First, four circRNAs, circ-5333-4, circ-0076490, circ-0047056, and circ-16293-1, were found to be dysregulated in the peripheral blood of MG patients. Among these, circ-5333-4 exhibited significant upregulation and was associated with QMGs, indicating a potential connection between its expression and MG severity ( 112 ). The specificity of circ-5333-4 for MG was highlighted by comparing MG and SLE cohorts. And circ-5333-4 was proposed to be part of a ceRNA regulation network involving miR-4310 and MORF4L2, warranting further investigation of potential for MG diagnosis and monitoring. Second, in a more recent study investigating circRNAs in MG serum, circ-FBL was identified as an overexpressed circRNA ( 113 ). It was suggested to function as a ceRNA by sponging miR-133, thereby promoting the expression of Pax7. This interaction was found to enhance myoblast proliferation, potentially compensating for MG-related muscle weakness, suggesting a therapeutic effect when Pax7 regulated by circ-FBL expressed to a great extent.
5 Conclusions and future directions
MG is characterized by complexity in diagnosis, treatment variability, and lacks curative options. ncRNAs have emerged as vital biomarkers for delineating MG subgroups, assessing severity, monitoring responses, and offering treatment targets. Research highlights aberrant ncRNAs in MG pathogenesis, suggesting their potential as biomarkers ( Table 4 ). While immune dysregulation and antibody production are known MG drivers, precise disease initiators remain elusive. ncRNAs are integral in MG pathogenesis, potentially offering avenues for disease prevention. Some differentially expressed ncRNAs could modulate T cell differentiation, disrupt the delicate balance between helper T cells and regulatory T cells, activate B cells, regulate inflammatory cytokine production, and function as ceRNAs or not to construct intricate networks involving related pathways. Moreover, Losen et al. ( 115 ) reported that short hairpin RNA (shRNA) could disturb the neuromuscular transmission by reducing the level of rapsyn, a bridge protein between AChR and the cytoskeleton in the postsynaptic membrane ( 2 ). In addition, some ncRNAs were reported to protect the muscle endplate from the complement attack ( 116 ). The regulation of several miRNAs, such as miR-206, miR-127, and miR-29b, can affect the differentiation of satellite cells, the main muscle stem cells ( 117 ). A few of ncRNAs were found to serve as ceRNAs to modulate the muscle development ( 113 , 117 ). It follows that ncRNAs may not only influence the production of specific antibodies, but also take part in the stabilization of postsynaptic membrane, the regulation of complement, and the regeneration of muscle.

Table 4 Sn, Sp, and AUC of ncRNAs as MG biomarker candidates.
Several drugs targeting T cells and B cells have sprung up to treat MG. In parallel, the burgeoning interest in the role of ncRNAs in modulating B cells and T cells presents a fertile ground for exploration. With the potential for regulating GCs formation, B cell differentiation, certain ncRNAs, such as miR-146a, miR-155, miR-139-5p, and miR-452-5p, exhibit potential as innovative therapeutic targets, while others may serve as prognostic indicators for clinical response. Furthermore, some ncRNAs with protective roles in MG may harbor therapeutic potential, like circ-FBL ( 113 ).
Beyond their therapeutic implications, ncRNAs can also serve as indicators for monitoring clinical therapeutic responsiveness, by changing in their levels before and after the treatment and effecting on antibody titers, thus facilitating individualized treatment approaches. Notably, the presence of some miRNAs, such as miR-323b-3p, miR-409-3p, miR-485-3p, miR-181d-5p and miR-340-3p, made patients insensitive to immunosuppressive therapies ( 47 ), suggesting the need for a reevaluation of treatment strategies.
The preceding findings underscore the significance of further exploration into the role of ncRNAs in MG. However, several critical areas remain uncharted. Firstly, a subset known as ‘seronegative MG’ ( 41 ) patients that lack detectable antibodies still present a diagnostic challenge, and the exploration of ncRNAs may offer insights into the diagnosis of them. Furthermore, we know less about the ncRNAs associated with uncommon antibodies at the neuromuscular junction, such as anti-LRP4 and Agrin antibodies. Secondly, many studies have focused solely on documenting the aberrant expression of ncRNAs without delving into their specific roles in MG pathogenesis. Lastly, there is a paucity of research on circRNAs in MG, despite their potential to uncover novel aspects of MG pathogenesis and open new avenues for treatment. In summary, the enigmatic pathogenesis of MG and its association with ncRNAs necessitate comprehensive and ongoing exploration to expand our understanding. This expanded knowledge will better equip us to navigate the complexities associated with the disease and develop more tailored and effective treatment approaches.
Author contributions
BW: Writing – review & editing, Writing – original draft. YZ: Writing – review & editing. DL: Writing – review & editing. CH: Writing – review & editing. RZ: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81501006, 81400950). The design collection and analysis were supported by the funding.
Acknowledgments
We are deeply grateful to all participants of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Huijbers MG, Marx A, Plomp JJ, Le Panse R, Phillips WD. Advances in the understanding of disease mechanisms of autoimmune neuromuscular junction disorders. Lancet Neurol . (2022) 21:163–75. doi: 10.1016/S1474-4422(21)00357-4
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Primers . (2019) 5:30. doi: 10.1038/s41572-019-0079-y
3. Punga AR, Maddison P, Heckmann JM, Guptill JT, Evoli A. Epidemiology, diagnostics, and biomarkers of autoimmune neuromuscular junction disorders. Lancet Neurol . (2022) 21:176–88. doi: 10.1016/S1474-4422(21)00297-0
4. Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia gravis: epidemiology, pathophysiology and clinical manifestations. J Clin Med . (2021) 10:2235. doi: 10.3390/jcm10112235
5. Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nat Rev Genet . (2010) 11:559–71. doi: 10.1038/nrg2814
6. Meller VH, Joshi SS, Deshpande N. Modulation of chromatin by noncoding RNA. Annu Rev Genet . (2015) 49:673–95. doi: 10.1146/annurev-genet-112414-055205
7. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature . (2013) 495:333–8. doi: 10.1038/nature11928
8. Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest . (2017) 127:761–71. doi: 10.1172/JCI84424
9. Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, et al. The emerging role of epigenetics in human autoimmune disorders. Clin Epigenet . (2019) 11:34. doi: 10.1186/s13148-019-0632-2
CrossRef Full Text | Google Scholar
10. Keijzers M, Nogales-Gadea G, de Baets M. Clinical and scientific aspects of acetylcholine receptor myasthenia gravis. Curr Opin Neurol . (2014) 27:552–7. doi: 10.1097/WCO.0000000000000125
11. Chen X, Yang T, Wang W, Xi W, Zhang T, Li Q, et al. Circular RNAs in immune responses and immune diseases. Theranostics . (2019) 9:588–607. doi: 10.7150/thno.29678
12. Schell SL, Rahman ZSM. miRNA-mediated control of B cell responses in immunity and SLE. Front Immunol . (2021) 12:683710. doi: 10.3389/fimmu.2021.683710
13. Zhang L, Wu H, Zhao M, Chang C, Lu Q. Clinical significance of miRNAs in autoimmunity. J Autoimmun . (2020) 109:102438. doi: 10.1016/j.jaut.2020.102438
14. Ali SA, Peffers MJ, Ormseth MJ, Jurisica I, Kapoor M. The non-coding RNA interactome in joint health and disease. Nat Rev Rheumatol . (2021) 17:692–705. doi: 10.1038/s41584-021-00687-y
15. Melzer N, Ruck T, Fuhr P, Gold R, Hohlfeld R, Marx A, et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society. J Neurol . (2016) 263:1473–94. doi: 10.1007/s00415-016-8045-z
16. Schneider-Gold C, Gilhus NE. Advances and challenges in the treatment of myasthenia gravis. Ther Adv Neurol Disord . (2021) 14:17562864211065406. doi: 10.1177/17562864211065406
17. Rodolico C, Nicocia G, Damato V, Antonini G, Liguori R, Evoli A. Benefit and danger from immunotherapy in myasthenia gravis. Neurol Sci . (2021) 42:1367–75. doi: 10.1007/s10072-021-05077-6
18. Catalanotto C, Cogoni C, Zardo G. MicroRNA in control of gene expression: an overview of nuclear functions. Int J Mol Sci . (2016) 17:1712. doi: 10.3390/ijms17101712
19. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell . (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
20. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell . (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
21. Chen JQ, Papp G, Szodoray P, Zeher M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev . (2016) 15:1171–80. doi: 10.1016/j.autrev.2016.09.003
22. Le Panse R, Berrih-Aknin S. Autoimmune myasthenia gravis: autoantibody mechanisms and new developments on immune regulation. Curr Opin Neurol . (2013) 26:569–76. doi: 10.1097/WCO.0b013e328364d6cd
23. Marcuse F, Brandts L, Moens D, Damoiseaux J, Hochstenbag M, Hoeijmakers JGJ, et al. The association between anti-acetylcholine receptor antibody level and clinical improvement in myasthenia gravis. Eur J Neurol . (2022) 29:1187–97. doi: 10.1111/ene.15238
24. Zhao R, Luo S, Zhao C. The role of innate immunity in myasthenia gravis. Autoimmun Rev . (2021) 20:102800. doi: 10.1016/j.autrev.2021.102800
25. Berrih-Aknin S, Le Panse R. Myasthenia gravis: a comprehensive review of immune dysregulation and etiological mechanisms. J Autoimmun . (2014) 52:90–100. doi: 10.1016/j.jaut.2013.12.011
26. Molin CJ, Sabre L, Weis CA, Punga T, Punga AR. Thymectomy lowers the myasthenia gravis biomarker miR-150-5p. Neurol Neuroimmunol Neuroinflamm . (2018) 5:e450. doi: 10.1212/NXI.0000000000000450
27. Qian K, Xu JX, Deng Y, Peng H, Peng J, Ou CM, et al. Signaling pathways of genetic variants and miRNAs in the pathogenesis of myasthenia gravis. Gland Surg . (2020) 9:1933–44. doi: 10.21037/gs
28. Yang L, Wang J, Sun X, Cao Y, Ning S, Zhang H, et al. Identifying a polymorphic ‘switch’ that influences miRNAs’ regulation of a myasthenia gravis risk pathway. PloS One . (2014) 9:e104827. doi: 10.1371/journal.pone.0104827
29. Sabre L, Maddison P, Wong SH, Sadalage G, Ambrose PA, Plant GT, et al. miR-30e-5p as predictor of generalization in ocular myasthenia gravis. Ann Clin Transl Neurol . (2019) 6:243–51. doi: 10.1002/acn3.692
30. Zhong H, Lu J, Jing S, Xi J, Yan C, Song J, et al. Low-dose rituximab lowers serum Exosomal miR-150-5p in AChR-positive refractory myasthenia gravis patients. J Neuroimmunol . (2020) 348:577383. doi: 10.1016/j.jneuroim.2020.577383
31. Lu H, Wang H, Sun P, Wang J, Li S, Xu T. MiR-522-3p inhibits proliferation and activation by regulating the expression of SLC31A1 in T cells. Cytotechnology . (2021) 73:483–96. doi: 10.1007/s10616-021-00472-5
32. Sabre L, Maddison P, Sadalage G, Ambrose PA, Punga AR. Circulating microRNA miR-21-5p, miR-150-5p and miR-30e-5p correlate with clinical status in late onset myasthenia gravis. J Neuroimmunol . (2018) 321:164–70. doi: 10.1016/j.jneuroim.2018.05.003
33. Xin Y, Cai H, Lu T, Zhang Y, Yang Y, Cui Y. miR-20b inhibits T cell proliferation and activation via NFAT signaling pathway in thymoma-associated myasthenia gravis. BioMed Res Int . (2016) 2016:9595718. doi: 10.1155/2016/9595718
34. Cron MA, Payet CA, Fayet OM, Maillard S, Truffault F, Fadel E, et al. Decreased expression of miR-29 family associated with autoimmune myasthenia gravis. J Neuroinflammat . (2020) 17:294. doi: 10.1186/s12974-020-01958-3
35. Punga T, Le Panse R, Andersson M, Truffault F, Berrih-Aknin S, Punga AR. Circulating miRNAs in myasthenia gravis: miR-150-5p as a new potential biomarker. Ann Clin Transl Neurol . (2014) 1:49–58. doi: 10.1002/acn3.24
36. Sabre L, Guptill JT, Russo M, Juel VC, Massey JM, Howard JF Jr, et al. Circulating microRNA plasma profile in MuSK+ myasthenia gravis. J Neuroimmunol . (2018) 325:87–91. doi: 10.1016/j.jneuroim.2018.10.003
37. Li J, Qiu D, Chen Z, Du W, Liu J, Mo X. Altered expression of miR-125a-5p in thymoma-associated myasthenia gravis and its down-regulation of foxp3 expression in Jurkat cells. Immunol Lett . (2016) 172:47–55. doi: 10.1016/j.imlet.2016.02.005
38. Zhang X, Liu S, Chang T, Xu J, Zhang C, Tian F, et al. Intrathymic tfh/B cells interaction leads to ectopic GCs formation and anti-AChR antibody production: central role in triggering MG occurrence. Mol Neurobiol . (2016) 53:120–31. doi: 10.1007/s12035-014-8985-1
39. Masuda M, Matsumoto M, Tanaka S, Nakajima K, Yamada N, Ido N, et al. Clinical implication of peripheral CD4+CD25+ regulatory T cells and Th17 cells in myasthenia gravis patients. J Neuroimmunol . (2010) 225:123–31. doi: 10.1016/j.jneuroim.2010.03.016
40. Li J, Qiu D, Chen Z, Du W, Liu J, Mo X. miR-548k regulates CXCL13 expression in myasthenia gravis patients with thymic hyperplasia and in Jurkat cells. J Neuroimmunol . (2018) 320:125–32. doi: 10.1016/j.jneuroim.2018.03.021
41. Lenti MV, Rossi CM, Melazzini F, Gastaldi M, Bugatti S, Rotondi M, et al. Seronegative autoimmune diseases: A challenging diagnosis. Autoimmun Rev . (2022) 21:103143. doi: 10.1016/j.autrev.2022.103143
42. Aricha R, Feferman T, Scott HS, Souroujon MC, Berrih-Aknin S, Fuchs S. The susceptibility of Aire (-/-) mice to experimental myasthenia gravis involves alterations in regulatory T cells. J Autoimmun . (2011) 36:16–24. doi: 10.1016/j.jaut.2010.09.007
43. McAllister E, Jellusova J. BAFF signaling in B cell metabolism. Curr Opin Immunol . (2021) 71:69–74. doi: 10.1016/j.coi.2021.05.011
44. Wang Z, Chen Y, Xu S, Yang Y, Wei D, Wang W, et al. Aberrant decrease of microRNA19b regulates TSLP expression and contributes to Th17 cells development in myasthenia gravis related thymomas. J Neuroimmunol . (2015) 288:34–9. doi: 10.1016/j.jneuroim.2015.08.013
45. Cheng Z, Qiu S, Jiang L, Zhang A, Bao W, Liu P, et al. MiR-320a is downregulated in patients with myasthenia gravis and modulates inflammatory cytokines production by targeting mitogen-activated protein kinase 1. J Clin Immunol . (2013) 33:567–76. doi: 10.1007/s10875-012-9834-5
46. Villegas JA, Van Wassenhove J, Le Panse R, Berrih-Aknin S, Dragin N. An imbalance between regulatory T cells and T helper 17 cells in acetylcholine receptor-positive myasthenia gravis patients. Ann N Y Acad Sci . (2018) 1413:154–62. doi: 10.1111/nyas.13591
47. Cavalcante P, Mizrachi T, Barzago C, Scandiffio L, Bortone F, Bonanno S, et al. MicroRNA signature associated with treatment response in myasthenia gravis: A further step towards precision medicine. Pharmacol Res . (2019) 148:104388. doi: 10.1016/j.phrs.2019.104388
48. Wang YZ, Tian FF, Yan M, Zhang JM, Liu Q, Lu JY, et al. Delivery of an miR155 inhibitor by anti-CD20 single-chain antibody into B cells reduces the acetylcholine receptor-specific autoantibodies and ameliorates experimental autoimmune myasthenia gravis. Clin Exp Immunol . (2014) 176:207–21. doi: 10.1111/cei.12265
49. Lu Y, Hippen KL, Lemire AL, Gu J, Wang W, Ni X, et al. miR-146b antagomir-treated human Tregs acquire increased GVHD inhibitory potency. Blood . (2016) 128:1424–35. doi: 10.1182/blood-2016-05-714535
50. Huang P, He XY, Xu M. Expression and significance of microRNA-126 and microRNA-21 in peripheral blood mononuclear cells in patients with myasthenia gravis. Neuroimmunomodulation . (2021) 3:1–8. doi: 10.1159/000510714
51. Howard JF Jr, Bril V, Burns TM, Mantegazza R, Bilinska M, Szczudlik A, et al. Efgartigimod MG Study Group. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology . (2019) 92:e2661–73. doi: 10.1212/WNL.0000000000007600
52. Punga AR, Andersson M, Alimohammadi M, Punga T. Disease specific signature of circulating miR-150-5p and miR-21-5p in myasthenia gravis patients. J Neurol Sci . (2015) 356:90–6. doi: 10.1016/j.jns.2015.06.019
53. Bo C, Zhang H, Cao Y, Lu X, Zhang C, Li S, et al. Construction of a TF-miRNA-gene feed-forward loop network predicts biomarkers and potential drugs for myasthenia gravis. Sci Rep . (2021) 11:2416. doi: 10.1038/s41598-021-81962-6
54. Sengupta M, Wang BD, Lee NH, Marx A, Kusner LL, Kaminski HJ. MicroRNA and mRNA expression associated with ectopic germinal centers in thymus of myasthenia gravis. PloS One . (2018) 13:e0205464. doi: 10.1371/journal.pone.0205464
55. Barzago C, Lum J, Cavalcante P, Srinivasan KG, Faggiani E, Camera G, et al. A novel infection- and inflammation-associated molecular signature in peripheral blood of myasthenia gravis patients. Immunobiology . (2016) 221:1227–36. doi: 10.1016/j.imbio.2016.06.012
56. Cao Y, Lu X, Wang J, Zhang H, Liu Z, Xu S, et al. Construction of an miRNA-regulated drug-pathway network reveals drug repurposing candidates for myasthenia gravis. Int J Mol Med . (2017) 39:268–78. doi: 10.3892/ijmm.2017.2853
57. Marx A, Yamada Y, Simon-Keller K, Schalke B, Willcox N, Ströbel P, et al. Thymus and autoimmunity. Semin Immunopathol . (2021) 43:45–64. doi: 10.1007/s00281-021-00842-3
58. Cron MA, Maillard S, Truffault F, Gualeni AV, Gloghini A, Fadel E, et al. Causes and consequences of miR-150-5p dysregulation in myasthenia gravis. Front Immunol . (2019) 10:539. doi: 10.3389/fimmu.2019.00539
59. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 directs FOXP3 localization and facilitates regulatory T-cell function through K63-linked ubiquitination. EMBO J . (2019) 38:e99766. doi: 10.15252/embj.201899766
60. Yan M, Fu Y, Rao H, Zhou H, Liang X. Expression and clinical significance of miR-146a and tumor necrosis factor receptor-associated factor 6 (TRAF6) in myasthenia gravis patient serum. BioMed Res Int . (2021) 2021:5573469. doi: 10.1155/2021/5573469
61. Cavalcante P, Bernasconi P, Mantegazza R. Autoimmune mechanisms in myasthenia gravis. Curr Opin Neurol . (2012) 25:621–9. doi: 10.1097/WCO.0b013e328357a829
62. Wang J, Zheng S, Xin N, Dou C, Fu L, Zhang X, et al. Identification of novel MicroRNA signatures linked to experimental autoimmune myasthenia gravis pathogenesis: down-regulated miR-145 promotes pathogenetic Th17 cell response. J Neuroimmune Pharmacol . (2013) 8:1287–302. doi: 10.1007/s11481-013-9498-9
63. Liu XF, Wang RQ, Hu B, Luo MC, Zeng QM, Zhou H, et al. MiR-15a contributes abnormal immune response in myasthenia gravis by targeting CXCL10. Clin Immunol . (2016) 164:106–13. doi: 10.1016/j.clim.2015.12.009
64. Lu J, Yan M, Wang Y, Zhang J, Yang H, Tian FF, et al. Altered expression of miR-146a in myasthenia gravis. Neurosci Lett . (2013) 555:85–90. doi: 10.1016/j.neulet.2013.09.014
65. Zhang J, Jia G, Liu Q, Hu J, Yan M, Yang B, et al. Silencing miR-146a influences B cells and ameliorates experimental autoimmune myasthenia gravis. Immunology . (2015) 144:56–67. doi: 10.1111/imm.12347
66. Bortone F, Scandiffio L, Marcuzzo S, Bonanno S, Frangiamore R, Motta T, et al. miR-146a in myasthenia gravis thymus bridges innate immunity with autoimmunity and is linked to therapeutic effects of corticosteroids. Front Immunol . (2020) 11:142. doi: 10.3389/fimmu.2020.00142
67. Wang Q, Liu Y, Kuang S, Li R, Weng N, Zhou Z. miR-181a ameliorates the progression of myasthenia gravis by regulating TRIM9. Evid Based Complement Alternat Med . (2021) 2021:1303375. doi: 10.1155/2021/1303375
68. Jiang L, Cheng Z, Qiu S, Que Z, Bao W, Jiang C, et al. Altered let-7 expression in Myasthenia gravis and let-7c mediated regulation of IL-10 by directly targeting IL-10 in Jurkat cells. Int Immunopharmacol . (2012) 14:217–23. doi: 10.1016/j.intimp.2012.07.003
69. Cron MA, Maillard S, Delisle F, Samson N, Truffault F, Foti M, et al. Analysis of microRNA expression in the thymus of Myasthenia Gravis patients opens new research avenues. Autoimmun Rev . (2018) 17:588–600. doi: 10.1016/j.autrev.2018.01.008
70. Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front Immunol . (2020) 11:212. doi: 10.3389/fimmu.2020.00212
71. Nogales-Gadea G, Ramos-Fransi A, Suárez-Calvet X, Navas M, Rojas-García R, Mosquera JL, et al. Analysis of serum miRNA profiles of myasthenia gravis patients. PloS One . (2014) 9:e91927. doi: 10.1371/journal.pone.0091927
72. Shi L, Liu T, Zhang M, Guo Y, Song C, Song D, et al. miR-15b is downregulated in myasthenia gravis patients and directly regulates the expression of interleukin-15 (IL-15) in experimental myasthenia gravis mice. Med Sci Monit . (2015) 21:1774–80. doi: 10.12659/MSM.893458
73. Xu H, Bao Z, Liang D, Li M, Wei M, Ge X, et al. Plasma exosomal miR-106a-5p expression in myasthenia gravis. Muscle Nerve . (2020) 61:401–7. doi: 10.1002/mus.26785
74. Zhang Y, Guo M, Xin N, Shao Z, Zhang X, Zhang Y, et al. Decreased microRNA miR-181c expression in peripheral blood mononuclear cells correlates with elevated serum levels of IL-7 and IL-17 in patients with myasthenia gravis. Clin Exp Med . (2016) 16:413–21. doi: 10.1007/s10238-015-0358-1
75. Chunjie N, Huijuan N, Zhao Y, Jianzhao W, Xiaojian Z. Disease-specific signature of serum miR-20b and its targets IL-8 and IL-25, in myasthenia gravis patients. Eur Cytokine Netw . (2015) 26:61–6. doi: 10.1684/ecn.2015.0367
76. Howard JF Jr, Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol . (2021) 20:526–36. doi: 10.1212/WNL.96.15_supplement.4520
77. Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, Batocchi AP, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain . (2003) 126:2304–11. doi: 10.1093/brain/awg223
78. Punga T, Bartoccioni E, Lewandowska M, Damato V, Evoli A, Punga AR. Disease specific enrichment of circulating let-7 family microRNA in MuSK+ myasthenia gravis. J Neuroimmunol . (2016) 292:21–6. doi: 10.1016/j.jneuroim.2016.01.003
79. Tan Y, Zhu L, Cui L, Guan Y. Differential expression of miRNA in the peripheral blood mononuclear cells in myasthenia gravis with muscle-specific receptor tyrosine kinase antibodies. Crit Rev Eukaryot Gene Expr . (2021) 31:1–15. doi: 10.1615/CritRevEukaryotGeneExpr.v31.i2
80. Bogatikov E, Lindblad I, Punga T, Punga AR. miR-1933-3p is upregulated in skeletal muscles of MuSK+ EAMG mice and affects Impa1 and Mrpl27. Neurosci Res . (2020) 151:46–52. doi: 10.1016/j.neures.2019.02.003
81. Zhou Y, Yan C, Gu X, Zhou L, Lu J, Zhu W, et al. Short-term effect of low-dose rituximab on myasthenia gravis with muscle-specific tyrosine kinase antibody. Muscle Nerve . (2021) 63:824–30. doi: 10.1002/mus.27233
82. Ma L, Pan L, Liu W, Liu Y, Xiang X, Pan Y, et al. Agrin Influences Botulinum Neurotoxin A-Induced Nerve Sprouting via miR-144-agrin-MuSK Signaling. Front Cell Dev Biol . (2020) 8:15. doi: 10.3389/fcell.2020.00015
83. Chaudhry N, Muhammad H, Seidl C, Downes D, Young DA, Hao Y, et al. Highly efficient CRISPR-Cas9-mediated editing identifies novel mechanosensitive microRNA-140 targets in primary human articular chondrocytes. Osteoarthritis Cartilage . (2022) 30:596–604. doi: 10.1016/j.joca.2022.01.005
84. Yu H, Zhang J, Liu X, Li Y. microRNA-136-5p from bone marrow mesenchymal stem cell-derived exosomes facilitates fracture healing by targeting LRP4 to activate the Wnt/β-catenin pathway. Bone Joint Res . (2021) 10:744–58. doi: 10.1302/2046-3758.1012.BJR-2020-0275.R2
85. Li SC, Shi H, Khan M, Caplin M, Meyer T, Öberg K, et al. Roles of miR-196a on gene regulation of neuroendocrine tumor cells. Mol Cell Endocrinol . (2015) 412:131–9. doi: 10.1016/j.mce.2015.06.003
86. Xie S, Li X, Qian L, Cai C, Xiao G, Jiang S, et al. An integrated analysis of mRNA and miRNA in skeletal muscle from myostatin-edited Meishan pigs. Genome . (2019) 62:305–15. doi: 10.1139/gen-2018-0110
87. Yu Z, Zhang M, Luo B, Jing H, Yu Y, Wang S, et al. Lrp4 in hippocampal astrocytes serves as a negative feedback factor in seizures. Cell Biosci . (2020) 10:135. doi: 10.1186/s13578-020-00498-w
88. Mao Z, Wang Z, Zhang S, Pu Y, Wang J, Zhang T, et al. LRP4 promotes migration and invasion of gastric cancer under the regulation of microRNA-140-5p. Cancer biomark . (2020) 29:245–53. doi: 10.3233/CBM-190571
89. Wang L, Zhang L. Emerging roles of dysregulated microRNAs in myasthenia gravis. Front Neurosci . (2020) 14:507. doi: 10.3389/fnins.2020.00507
90. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell . (2011) 43:904–14. doi: 10.1016/j.molcel.2011.08.018
91. Noh JH, Kim KM, McClusky WG, Abdelmohsen K, Gorospe M. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip Rev RNA . (2018) 9:e1471. doi: 10.1002/wrna.1471
92. Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol . (2017) 18:962–72. doi: 10.1038/ni.3771
93. Hong Y, Liang X, Gilhus NE. AChR antibodies show a complex interaction with human skeletal muscle cells in a transcriptomic study. Sci Rep . (2020) 10:11230. doi: 10.1038/s41598-020-68185-x
94. Hu B, Niu L, Jiang Z, Xu S, Hu Y, Cao K. LncRNA XLOC_003810 promotes T cell activation and inhibits PD-1/PD-L1 expression in patients with myasthenia gravis-related thymoma. Scand J Immunol . (2020) 92:e12886. doi: 10.1111/sji.12886
95. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell . (2011) 146:353–8. doi: 10.1016/j.cell.2011.07.014
96. Zhang F, Liu G, Bu Y, Ma X, Hao J. Expression profile of long noncoding RNAs and mRNAs in peripheral blood mononuclear cells from myasthenia gravis patients. J Neuroimmunol . (2016) 299:124–9. doi: 10.1016/j.jneuroim.2016.09.005
97. Lu W, Lu Y, Wang CF, Chen TT. Expression profiling and bioinformatics analysis of exosomal long noncoding RNAs in patients with myasthenia gravis by RNA sequencing. J Clin Lab Anal . (2021) 35:e23722. doi: 10.1002/jcla.23722
98. Xu Y, Ouyang Y. Long non-coding RNA growth arrest specific 5 regulates the T helper 17/regulatory T balance by targeting miR-23a in myasthenia gravis. J Int Med Res . (2022) 50:3000605211053703. doi: 10.1177/03000605211053703
99. Li S, Wang X, Wang T, Zhang H, Lu X, Liu L, et al. Identification of the regulatory role of lncRNA HCG18 in myasthenia gravis by integrated bioinformatics and experimental analyses. J Transl Med . (2021) 19:468. doi: 10.1186/s12967-021-03138-0
100. Luo M, Liu X, Meng H, Xu L, Li Y, Li Z, et al. IFNA-AS1 regulates CD4+ T cell activation in myasthenia gravis though HLA-DRB1. Clin Immunol . (2017) 183:121–31. doi: 10.1016/j.clim.2017.08.008
101. Wang J, Cao Y, Lu X, Wang X, Kong X, Bo C, et al. Identification of the regulatory role of lncRNA SNHG16 in myasthenia gravis by constructing a competing endogenous RNA network. Mol Ther Nucleic Acids . (2020) 19:1123–33. doi: 10.1016/j.omtn.2020.01.005
102. Niu L, Jiang J, Yin Y, Hu B. LncRNA XLOC_003810 modulates thymic Th17/Treg balance in myasthenia gravis with thymoma. Clin Exp Pharmacol Physiol . (2020) 47:989–96. doi: 10.1111/1440-1681.13280
103. Peng S, Huang Y. LncRNA GAS5 positively regulates IL-10 expression in patients with generalized myasthenia gravis. Brain Behav . (2022) 12:e2457. doi: 10.1002/brb3.2457
104. Xu S, Wang T, Lu X, Zhang H, Liu L, Kong X, et al. Identification of LINC00173 in myasthenia gravis by integration analysis of aberrantly methylated- differentially expressed genes and ceRNA networks. Front Genet . (2021) 12:726751. doi: 10.3389/fgene.2021.726751
105. Wang X, Zhang H, Lu X, Li S, Kong X, Liu L, et al. LncRNA OIP5-AS1 modulates the proliferation and apoptosis of Jurkat cells by sponging miR-181c-5p to regulate IL-7 expression in myasthenia gravis. PeerJ . (2022) 10:e13454. doi: 10.7717/peerj.13454
106. Ke J, Du X, Cui J, Yu L, Li H. LncRNA and mRNA expression associated with myasthenia gravis in patients with thymoma. Thorac Canc . (2022) 13:15–23. doi: 10.1111/1759-7714.14201
107. Luo Z, Li Y, Liu X, Luo M, Xu L, Luo Y, et al. Systems biology of myasthenia gravis, integration of aberrant lncRNA and mRNA expression changes. BMC Med Genomics . (2015) 8:13. doi: 10.1186/s12920-015-0087-z
108. Kong X, Wang J, Cao Y, Zhang H, Lu X, Wang Y, et al. The long noncoding RNA MALAT-1 functions as a competing endogenous RNA to regulate MSL2 expression by sponging miR-338-3p in myasthenia gravis. J Cell Biochem . (2019) 120:5542–50. doi: 10.1002/jcb.27838
109. Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A . (1976) 73:3852–6. doi: 10.1073/pnas.73.11.3852
110. Chen B, Huang S. Circular RNA: An emerging non-coding RNA as a regulator and biomarker in cancer. Cancer Lett . (2018) 418:41–50. doi: 10.1016/j.canlet.2018.01.011
111. Xie R, Zhang Y, Zhang J, Li J, Zhou X. The role of circular RNAs in immune-related diseases. Front Immunol . (2020) 11:545. doi: 10.3389/fimmu.2020.00545
112. Lv J, Ren L, Han S, Zhang J, Zhao X, Zhang Y, et al. Peripheral blood hsa-circRNA5333-4: A novel biomarker for myasthenia gravis. Clin Immunol . (2021) 224:108676. doi: 10.1016/j.clim.2021.108676
113. Lai X, Bi Z, Yang X, Hu R, Wang L, Jin M, et al. Upregulation of circ-FBL promotes myogenic proliferation in myasthenia gravis by regulation of miR-133/PAX7. Cell Biol Int . (2021) 45:2287–93. doi: 10.1002/cbin.11676
114. Beretta F, Huang YF, Punga AR. Towards Personalized Medicine in Myasthenia Gravis: Role of Circulating microRNAs miR-30e-5p, miR-150-5p and miR-21-5p. Cells . (2022) 11:740. doi: 10.3390/cells11040740
115. Martínez-Martínez P, Phernambucq M, Steinbusch L, Schaeffer L, Berrih-Aknin S, Duimel H, et al. Silencing rapsyn in vivo decreases acetylcholine receptors and augments sodium channels and secondary postsynaptic membrane folding. Neurobiol Dis . (2009) 35:14–23. doi: 10.1016/j.nbd.2009.03.008
116. Kaminski HJ, Li Z, Richmonds C, Lin F, Medof ME. Complement regulators in extraocular muscle and experimental autoimmune myasthenia gravis. Exp Neurol . (2004) 189:333–42. doi: 10.1016/j.expneurol.2004.06.005
117. Buonaiuto G, Desideri F, Taliani V, Ballarino M. Muscle regeneration and RNA: new perspectives for ancient molecules. Cells . (2021) 10:2512. doi: 10.3390/cells10102512
Keywords: myasthenia gravis, non-coding RNAs, pathogenesis, treatment, biomarker
Citation: Wang B, Zhu Y, Liu D, Hu C and Zhu R (2024) The intricate dance of non-coding RNAs in myasthenia gravis pathogenesis and treatment. Front. Immunol. 15:1342213. doi: 10.3389/fimmu.2024.1342213
Received: 21 November 2023; Accepted: 11 March 2024; Published: 27 March 2024.
Reviewed by:
Copyright © 2024 Wang, Zhu, Liu, Hu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruixia Zhu, [email protected]
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Front Neurol
A Practical Approach to Managing Patients With Myasthenia Gravis—Opinions and a Review of the Literature
Maria elena farrugia.
1 Neurology Department, Institute of Neurological Sciences, Queen Elizabeth University Hospital, Glasgow, United Kingdom
John A. Goodfellow
2 Neuroimmunology Laboratory, Laboratory Medicine and Facilities Building, Queen Elizabeth University Hospital, Glasgow, United Kingdom
When the diagnosis of myasthenia gravis (MG) has been secured, the aim of management should be prompt symptom control and the induction of remission or minimal manifestations. Symptom control, with acetylcholinesterase inhibitors such as pyridostigmine, is commonly employed. This may be sufficient in mild disease. There is no single universally accepted treatment regimen. Corticosteroids are the mainstay of immunosuppressive treatment in patients with more than mild MG to induce remission. Immunosuppressive therapies, such as azathioprine are prescribed in addition to but sometimes instead of corticosteroids when background comorbidities preclude or restrict the use of steroids. Rituximab has a role in refractory MG, while plasmapheresis and immunoglobulin therapy are commonly prescribed to treat MG crisis and in some cases of refractory MG. Data from the MGTX trial showed clear evidence that thymectomy is beneficial in patients with acetylcholine receptor (AChR) antibody positive generalized MG, up to the age of 65 years. Minimally invasive thymectomy surgery including robotic-assisted thymectomy surgery has further revolutionized thymectomy and the management of MG. Ocular MG is not life-threatening but can be significantly disabling when diplopia is persistent. There is evidence to support early treatment with corticosteroids when ocular motility is abnormal and fails to respond to symptomatic treatment. Treatment needs to be individualized in the older age-group depending on specific comorbidities. In the younger age-groups, particularly in women, consideration must be given to the potential teratogenicity of certain therapies. Novel therapies are being developed and trialed, including ones that inhibit complement-induced immunological pathways or interfere with antibody-recycling pathways. Fatigue is common in MG and should be duly identified from fatigable weakness and managed with a combination of physical therapy with or without psychological support. MG patients may also develop dysfunctional breathing and the necessary respiratory physiotherapy techniques need to be implemented to alleviate the patient's symptoms of dyspnoea. In this review, we discuss various facets of myasthenia management in adults with ocular and generalized disease, including some practical approaches and our personal opinions based on our experience.
Introduction
Myasthenia gravis (MG) is a rare acquired autoimmune disorder of the neuromuscular junction (NMJ), caused by antibodies that target the post-synaptic membrane ( 1 ). These antibodies commonly are to the nicotinic acetylcholine receptor (AChR) but in a smaller proportion of cases, antibodies to muscle specific tyrosine kinase (MuSK) or to lipoprotein receptor-related protein 4 (Lrp-4) can be present instead ( 1 – 3 ). In an even smaller cohort of MG patients, no antibodies are detected on conventional antibody assay testing and we refer to these patients as “seronegative.” Patients with MG typically present with fatigable muscle weakness. They commonly present first with ocular manifestations such as asymmetrical fatigable ptosis with or without blurred or double vision. The majority, however, evolve further into generalized muscle weakness involving the facial and bulbar muscles, the neck and axial muscles and the limbs, with the upper limbs often being more severely affected than the lower limbs. In myasthenic crisis, the severe end of the disease spectrum, there is neuromuscular dysphagia rapidly evolving into complete loss of swallow function, and often in association with respiratory muscle weakness and type 2 respiratory failure. This is a clinical emergency that requires management in an intensive care setting. Therapies in the field of MG have significantly advanced over the years. Now, more than ever, the treating physician must carefully contemplate which treatments are best suited for an individual MG patient since the “one size fits all” approach may not be as relevant. There are specific clinical scenarios where one must be extra cautious, for instance the newly diagnosed young female patient, who may be imminently planning a pregnancy, in contrast to a newly diagnosed elderly patient with multiple comorbidities. This review discusses the literature with some emphasis on our practice based over a time-span of over a decade where we have treated an excess of 900 MG patients.
Pharmacological Therapies in Generalized MG
Medical therapies are used in MG patients for either direct alleviation of symptoms, or as immunomodulatory drugs with the aim of dampening the underlying immunopathology causing the disease. The aim of treatment is to induce remission (pharmacological in the majority or complete stable remission which is rarely achieved) or minimal manifestations (MM). The Myasthenia gravis Foundation of America (MGFA) post-intervention status (PIS) ( 4 ) defines MM in a patient who has no symptoms or functional limitations from MG but has some weakness on examination of some muscles. There are four different categories of MM depending on whether the patient is receiving treatment and if this includes immunosuppression and/or symptomatic treatment (for example pyridostigmine as will be discussed below). This contrasts to complete stable remission (CSR) where the patient has no symptoms of MG and no weakness (excluding residual weakness of eye closure) and has received no therapy for a minimum period of 1 year, and pharmacological remission (PR) which is the same as CSR but the patient would have received some therapy for MG excluding symptomatic treatment.
Symptomatic Therapies
Pyridostigmine is by far the most commonly used symptomatic therapy. This is an acetylcholinesterase inhibitor which blocks the degradation of acetylcholine at peripheral cholinergic synapses, including the neuromuscular junction (NMJ). Originally, physostigmine and prostigmine (neostigmine) were identified by Mary Broadfoot Walker, a physician in Scotland in the late 1880s, as drugs that temporarily improved muscle strength in patients with MG ( 5 ). These drugs work by prolonging the action of any acetylcholine released into the synaptic cleft and compensates for the structural and functional deficits in NMJ transmission that characterizes MG. In early or mild disease pyridostigmine allows significant and rapid improvement in muscle strength ( 6 , 7 ). However, with longstanding or severe disease this pharmacological compensation may be insufficient and there may be minimal clinical effect. Peak blood levels of pyridostigmine occur 1.5–3 h after oral intake but significant clinical effect occurs within 30 min. Dosing 4–5 times per day leads to very stable blood levels. Renal impairment leads to reduced clearance of pyridostigmine and doses must be adjusted.
Patients are usually prescribed doses of 180–240 mg daily but patients may require up to 480 mg daily. Although generally well-tolerated, side effects from pyridostigmine are very common, are usually dose dependent, and can be debilitating necessitating reduction of dose or slower titration. Most side effects arise from the action of pyridostigmine at non-NMJ muscarinic peripheral synapses and include, gastrointestinal disturbance (abdominal cramps, bloating, diarrhea, frequency, nausea), urinary frequency, hypotension, bradycardia, sweating, salivation, lacrimation, increased bronchial secretions, and other symptoms of cholinergic excess. Some elderly patients can be extremely sensitive to the cardiac side effects and have experienced syncope even with low doses of pyridostigmine. Some asthmatic patients may show increased sensitivity and experience increased bronchospasm with pyridostigmine. At high doses side effects can be severe, and lead to the entity of “cholinergic crisis,” where neuromuscular weakness worsens along with the above symptoms leading to bulbar or respiratory crisis from drug excess rather than worsening MG ( 8 ). Such extreme manifestations are uncommon but it is very frequent for patients to have gastrointestinal symptoms on starting or increasing doses. These tend to lessen within a few days but can persist in some. Propantheline is an antimuscarinic agent that counteracts many of the cholinergic side effects of pyridostigmine without reducing its action at the NMJ. It can be very effective at reducing the side effects of pyridostigmine if given ~15 min beforehand. Loperamide can alternatively be prescribed but is not as effective at reducing the other muscarinic side effects. When patients fail to respond to pyridostigmine, the physician should be cautious about increasing the dose particularly in dysphagic patients, since pyridostimgine will increase salivary secretions and exacerbate their swallowing difficulties.
Neostigmine is an alternative acetylcholine esterase inhibitor that can be used in MG ( 9 , 10 ). This should only be given via the subcutaneous route in MG and not intravenously. It is useful in patients with MG who cannot absorb via the oral route (e.g., a MG patient with acute bowel obstruction) but should not be first line if the patient has impaired swallow. Swallowing difficulties are very common in patients with MG and if there are concerns about aspiration with oral intake, including medications, the first strategy should always be to place a nasogastric tube and administer pyridostigmine via this. Only if this cannot be undertaken should subcutaneous neostigmine be used. It has the same side effect profile as pyridostigmine albeit with more marked cardioinhibitory effects and a shorter half-life leading to more frequent dosing. However, neostigmine should always be used with caution since it may cause excessive salivary secretions and as a result may further negatively impact and exacerbate swallowing difficulties.
Experimental models of AChR deficiency show how oral ß-2 adrenergic receptor agonists such as salbutamol enhance function of the NMJ ( 11 ). Oral salbutamol can rarely be of clinical utility in mild autoimmune MG disease too especially where the patient has not tolerated pyridostigmine. We have used successfully in a couple of patients. Side effects commonly include tachycardia, tremor and a sense of anxiety and these can be limiting factors. MG patients with MuSK antibodies tolerate albuterol and 3,4-diaminopyridine ( 12 ) more than pyridostigmine which, in MuSK-MG, is commonly associated with enhanced side effects especially of cramp and muscle fasciculations. A small clinical trial (phase IIB) studying amifampridine phosphate in MuSK-MG demonstrated this drug to be safe and effective ( 13 ). Ephedrine, a sympathomimetic agent, can also be used as an add-on treatment and improves symptoms and weakness ( 14 ). Tirasemtiv has been explored in a clinical trial and found to increase the muscle response to calcium and improves muscle strength in MG ( 15 ). This remains an experimental drug.
Immunomodulatory Therapies for Generalized MG
Corticosteroids.
Prednisolone or prednisone constitute the main immunomodulatory therapy in the long-term management of patients with MG ( 16 , 17 ). The majority will require long-term oral corticosteroid therapy and it is crucial to have the appropriate discussion with newly diagnosed patients, indicating that this will not be a short course of treatment. It is equally important to discuss with patients the long list of potential side effects from steroids, necessitating bone and gastric protection. Patients should also be adequately monitored for the development of diabetes mellitus, hypertension, with careful counseling on potential excessive weight gain and the necessary dietary changes that they may need to pre-emptively and pro-actively address. Other side effects include the formation of cataracts, raised intraocular pressures, mood and sleep disturbances, peripheral oedema and susceptibility to frequent infections or even sepsis. The latter may result in failure of response to conventional MG therapies or even a chronic refractory state and decline in status with multiple hospital admissions.
There can be a paradoxical worsening of MG symptoms on commencing corticosteroids at high doses ( 18 ). Therefore, our practice is to start at a low dose and escalate the dose gradually ( 16 ). Our initial practice was to use an alternate day regimen of steroids, where side effects are probably reduced when compared to the daily dosing schedule. However, we have encountered many difficulties with the alternate day regimen including patients and physicians in primary and secondary care becoming easily confused, and we have therefore resorted, in the last 3 years or so, to applying the daily steroid regimen. We initiate prednisolone at 5 mg daily and increase every third dose (day) by 5 mg until we achieve stability in MG symptoms and significant improvement, with our ceiling dose usually being 50 mg daily but higher doses have been prescribed in a few select cases.
We treat the majority of patients in the outpatient setting, giving clear instructions to the primary care physician and to the patient, with contact details of the myasthenia team. The nurse specialist phones in on the patient regularly to ensure that the treatment plan is being ensued and to monitor patients' symptoms over the phone. In patients demonstrating significant bulbar weakness, our preference is to admit them immediately to the neurology ward and to initiate treatment accordingly including symptomatic treatment with pyridostigmine and where necessary intravenous immunoglobulin (ivIG).
With the slow steroid dose escalation that we apply, patients improve after 2–4 months of initiation, but some do take much longer to improve significantly. This can be problematic in some, and occurs in circa 20% of patients that we manage. In patients with moderate bulbar muscle involvement or disabling fatigable limb weakness, we prefer to admit to the acute neurology ward or to the day-case ward (if they are generally stable) to treat them with a course of ivIG during the steroid escalation process in order to help expedite the process of their recovery. Occasionally, patients require more than a single course of ivIG to help stabilize their symptoms or to significantly improve their symptoms while increasing their corticosteroid dose. Some patients may not respond to ivIG. In this case, we employ plasma exchange (PE) if we feel their symptoms are sufficiently disabling. If patients are stable (but symptomatic) then PE can be administered in a day-case unit in an outpatient setting and PE carried out through peripheral venous access.
The slow steroid escalation regimen of treatment that we employ is in contrast to the early fast-acting treatment (EFT) strategies applied by the Japanese group ( 19 , 20 ). This strategy always involves patients being admitted to hospital for treatment where they would receive 1–2 plasmapheresis sessions followed immediately by high-dose intravenous methylprednisolone (0.5–1 g), with or without intravenous immunoglobulin therapy. Treatment would be repeated if significant improvement did not take place. Patients were then discharged from hospital on the lowest dose possible of oral steroids. In some patients, who did not have severe MG symptoms, high dose methylprednisolone was not required. Achievement of MM was more frequent and occurred earlier in the EFT therapy cohort were compared to those in the non-EFT one ( 19 , 20 ). While this regimen of treatment is highly attractive, it does require easy access to neurology inpatient beds and the necessary manpower (for instance accessibility to the plasmapheresis team) and would not be practical in our regional neurology center (which has 21 neurology beds serving a population of 2 million).
Steroid Sparing Immunosuppressive Agents
Until recently our practice has been to initiate a steroid sparing agent such as azathioprine, almost simultaneously as initiating corticosteroids and using a dose of 2.5 mg/kg/day ( 17 ). This was based on the study by Palace et al. ( 21 ) which showed that azathioprine was an effective adjunct treatment to prednisolone and was effective in reducing the long-term maintenance prednisolone dose, in reducing relapses, and in achieving remission in the long-term. However, our practice changed a few years ago ( 16 , 22 ), when we began to treat newly-diagnosed MG patients with steroids alone first. A steroid-sparing immunosuppressive agent would be later added if the patient relapsed while reducing their steroid dose indicating that they will require more than 10 mg daily of prednisolone to maintain MM and thus justifying the addition of such an agent. We also consider adding in immunosuppression early if the patient has pre-existing comorbidities such as diabetes, significant depression (with steroids potentially exacerbating their mood), osteoporosis, leg ulcerations, that would be compounded by several-month treatment with corticosteroids. Also, in patients who are demonstrating a slow response with corticosteroid treatment then we would an immunosuppressant early in the course of treatment. Furthermore, in some patients, corticosteroid treatment is absolutely or relatively contraindicated because of background comorbidities and in this scenario we immediately prescribe a steroid-sparing immunosuppressant agent without the addition of steroids. Stabilization can be prolonged with this strategy, and we prescribe ivIG in the interim with or without low-dose corticosteroids depending on the clinical picture. Some patients refuse to be started on steroids because of concerns of side effects and in these circumstances adding a steroid-sparing immunosuppressant at diagnosis is a viable option. A retrospective study by Abuzinadah et al. ( 23 ), showed that a satisfactory response (which included CSR, PR, and MM) was achieved in about 50% of MG patients with generalized disease when they were maintained on low dose prednisolone, without a steroid-sparing immunosuppressant with follow-up extending up to 6 years.
We advocate checking thiopurine S-methyltransferase (TPMT) levels ( 24 ) prior to initiating azathioprine treatment. If levels are in the normal range, we initiate azathioprine at 25 mg daily and increase weekly by 25 mg until target dose is reached, with blood monitoring carried out in primary practice. Generally, the drug is well-tolerated and we rarely encounter idiosyncratic reactions in our population. The drug however takes 8–12 months to become effective and we counsel patients about this. In our opinion the drug is not entirely benign and we have observed many patients develop multiple skin lesions namely actinic keratosis, as a result of long-term azathioprine use and also skin malignancies such as squamous cell carcinoma. If the TPMT levels are deficient but not absent, then we consider using lower doses of azathioprine, monitoring the level of the active metabolite, 6-thioguanine nucleotides (6-TGN), in the blood and titrating the dose accordingly.
Our second steroid-sparing agent of choice is mycophenolate mofetil (MMF) at a dose of 1 g twice daily. In general, we have found it practical to use this drug and it is well-tolerated and (as previously reported in the literature) ( 25 , 26 ) except for a small number of patients who complain of associated side effects including disabling dizziness and insomnia, and have discontinued this as a result. In patients with very high body mass indices, we have used doses of up to 2.5 g daily. Infrequently we have prescribed mycophenolic acid which can be better tolerated than MMF, in those with side effects from MMF. We find that the efficacy of MMF is noted after circa 6 months of treatment as was also observed in previous studies ( 27 ). Based on our clinical observations, and in contrast to the findings from a previous randomized controlled trial ( 28 ) oral weekly methotrexate is as effective as MMF and its efficacy becomes apparent at around the same time-point as MMF. It is about 20 times cheaper than MMF. Nausea and vomiting can be limiting side effects experienced by some. In general, folic acid 5 mg daily is prescribed day 4 after methotrexate but when nausea is prominent, daily folic acid (except for the day of methotrexate dosing) can help alleviate this. Ciclosporin (used at a dose of 3.5 mg/kg/day) is probably the most potent immunosuppressive agent with the added advantage that it is not teratogenic ( 29 ). From our clinical observations, we have deduced that ciclosporin is, at minimum, effective within 3 months of initiation. However, we have observed that the majority of patients prescribed this drug run into problems with significant side effects including hypertension, alteration in their glomerular filtration rates, nephrotoxicity, tremor and in female patients also problems with hirsutism. We have prescribed ciclosporin in around 25 MG patients, where they have proven refractory to other steroid-sparing immunosuppressants, and usually belonging to a younger age-group. We avoid prescribing in older patients because of the potential complications and side effects and aim to reserve for younger patient groups. Tacrolimus is of similar efficacy ( 30 ) but with a similar side effect profile as ciclosporin. We have not prescribed cyclophosphamide in MG but there is a role for prescribing this drug as a monthly intravenous pulsed treatment in patients with refractory disease and who are unable to reduce their maintenance steroid doses ( 31 ), and this is generally tolerated without significant side effects.
Withdrawing Symptomatic Therapies and Achieving Maintenance Therapy
When the MG status starts to stabilize, MG patients no longer experience the significant fluctuation and variability in symptoms, become less fatigable and their strength starts to normalize. We educate patients about this time-point being reached and trying to recognize when they no longer need to reach out for their next pyridostigmine dose which is a good prognostic sign for stabilization. At that stage, while maintaining the same dose of corticosteroids, we advise patients to reduce their pyridostigmine dose by 30 mg per week (or sometimes faster), with the aim to wean this altogether but in some cases patients prefer or require to remain on low doses of up to 120 mg daily. Reduction of their steroid dose then ensues following a similar regimen previously described ( 16 ) −5 mg reduction per month down to 20 mg daily, then 2.5 mg reductions per month down to 10 mg daily, then 1 mg reduction per month or slower, aiming to reach 5 mg daily. In some cases, it is possible to wean steroids altogether especially if a steroid sparing immunosuppressive agent has already been added. However, if this is not the case then careful consideration needs to be taken, with detailed discussion with the patient, about withdrawing steroids altogether and a potential risk of future relapse. There is an argument for maintaining on low-dose prednisolone such as 5 mg daily for life where the cumulative life-time risk is likely to be small vs. further reduction or absolute withdrawal of prednisolone that might trigger a significant relapse of MG. In our experience, most patients favor the former option. Also, we are of the opinion that the long-term risk of such low-dose prednisolone (development of diabetes, hypertension, osteoporosis, glaucoma etc.) is significantly less than for example being maintained on 100 mg of azathioprine for life—although there are no long-term studies that quantitate this risk.
Reducing the Dose of Second-Line Agents
When patients have achieved pharmacological remission and have successfully withdrawn corticosteroids, then it would be sensible to consider a gentle reduction in their steroid sparing immunosuppressant dose ( 17 ). The difficulties are 2-fold: firstly there is little data on the actual risk of relapse on withdrawal of immunosuppression and secondly there is no consensus or guideline on how rapidly the dose should be reduced. With regards to the first point, the limited studies on this indicate that the risk of relapse on withdrawal of immunosuppression may be rather high. In two respective studies, more than 50% of patients who were in CSR and who were prescribed azathioprine ( 32 ) and nearly all patients who had significantly reduced the dose or withdrawn MMF, experienced a relapse in their MG ( 33 ) necessitating the reintroduction of immunosuppression. With regards to the second difficulty: we usually take an ultra-conservative approach when reducing the dose of any immunsuppressant. In the case of azathioprine we reduce the dose by 25 mg every 6 months (infrequently weaning altogether) while with MMF we reduce no faster by 500 mg per year, as previously reported ( 34 ). We always advocate close monitoring of patients' MG status and symptoms during the reduction process. The rate of CSR is low and we often opt, after discussion with patients and balancing the decision against their age and comorbidities, to maintain them on the lowest dose possible of immunosuppressant in the long-term unless there is a pressing requirement that this is discontinued altogether.
Thymectomy in generalized AChR antibody positive MG should be considered as early as possible in the management plan and thymectomy should be performed where relevant when the MG status has been stabilized ( 17 ). Imaging of the thymus gland, using CT or MR modalities, should be performed in all AChR antibody positive MG patients, also to rule out thymoma and in the younger patients to look for evidence thymic hyperplasia. The role of the thymus gland in driving MG has been known for almost a century ( 35 , 36 ). The results from the international thymectomy trial (MGTX) have been crucial in underscoring the role of thymectomy in the management of MG ( 37 ). In this trial, non-thymoma MG patients up to the age of 65, with generalized disease and with positive AChR antibodies, were recruited. Patients whose MG onset was up to 5 years prior were recruited. The goal of the surgical procedure, in those who received thymectomy, was to remove all thymic tissue including ectopic tissue and surrounding fat. The results showed that patients who had thymectomy (which involved an extended trans-sternal procedure) required lesser doses of corticosteroids both in the short and in the long-term ( 38 ), had better functional outcomes, were less likely to be hospitalized due to their MG and were less likely to require additional immunosuppression with azathioprine for instance. The benefit was seen across all age-groups and was sustained on follow-up. This trial has been pivotal in the way we neurologists are now approaching MG management. Thymectomy now is more widely offered to patients with generalized disease associated with AChR antibodies, including patients with late-onset MG and up to the age of 65, as part of the overall treatment of their MG.
Minimally-invasive thymectomy surgery has been further revolutionary in the field. Reports of video-assisted thorascopic surgery (VATS) thymectomy began to emerge in 1993 and 1994, with a number of centers using alone or in combination with a trans-cervical approach ( 39 – 41 ). Reports of robotic surgery in the field of thoracic surgery began to emerge in the early 2000s ( 42 ) with the use of the da Vinci robotic system applied in a 28-year old patient with MG. With both types of minimally invasive procedures, patients are reported to experience less blood loss intra-operatively, complain of less pain post-operatively and have a shorter post-operative hospital stay when compared to open thymectomy. In a systemic review comparing robotic assisted thymectomy surgery (RATS) with VATS and open surgery ( 43 ), there are clear advantages of RATS or VATS over open surgery, but no significant advantage of RATS over VATS at least to date. Clinical outcomes have been compared in a retrospective study in MG and found to be comparable between thoracoscopic vs. trans-sternal thymecetomy ( 44 ). Data analyses, after propensity score matching, also confirmed that robotic thymectomy in early stage thymoma was safe and feasible with oncological outcomes that were comparable to trans-sternal thymectomy ( 45 ) and in thymoma exceeding 5 cm ( 46 , 47 ). In relation to MG outcomes, it would be very challenging to design a further trial that would compare clinical MG status and outcomes after open thymectomy vs. minimally invasive surgery.
In our experience, patients with non-thymoma MG are now more encouraged to pursue thymectomy, during the course of their MG management, when provided with the results from the MGTX trial. We have also observed that patients are also more comfortable in pursuing minimally invasive thymectomy surgery in contrast to open surgical approaches. For the past 3 years, our thoracic surgeons have been employing RATS, which we perceive as further advantageous specifically from the perspective of post-operative morbidity. For those who are in employment, who drive, who are parents looking after young children, and also for those younger adults who may be pursuing studies at school or university, RATS evokes less anxiety about the post-operative period impacting on their work, studies, social, or family life. Minimally invasive thymectomy procedures also overcome the aesthetic problems that patients faced with open thymectomy mediastinal scars. We, as a center, have also gained confidence in referring older MG patients for thymectomy, acknowledging that there is data to support its benefit also in this age-group ( 48 , 49 ), and since the MGTX trial, we have been consistently referring patients up to the age of 65.
Although it is perceived that that there is a 2-year window of opportunity for thymectomy from disease onset, there is no evidence to suggest that the MG status is negatively impacted when thymectomy is performed beyond this time-frame. In the MGTX trial there was no evidence to support that patients who had thymectomy within 2 years did better than those who had thymectomy within 5 years of disease onset. This is particularly relevant to patients, who have proven refractory to all conventional immunosuppression, and where thymectomy at a later time-point in their disease could potentially offer additional benefit; we have been exploring this as an option in a small category of patients. In contrast, there are various reports indicating that thymectomy is contraindicated in MuSK-MG, with patients' MG status often worsening after the procedure and, therefore, thymectomy should not be pursued if MuSK antibodies are present ( 50 ). The jury is out as to whether thymectomy would benefit MG patients without AChR or MuSK antibodies (traditionally referred to as double seronegative) and if there is a role for thymectomy in MG with LRP-4 antibodies ( 51 ). Leite et al. ( 52 ) had shown that the thymic abnormalities in double seronegative patients had more thymic abnormalities than the MuSK-MG thymus, but less than seen in the AChR-MG cohort ( 52 ). Thus, these patients may benefit from thymectomy too but this is an area that requires further research.
Thymoma, in contrast, is a rare epithelial tumor of the anterior mediastinum and 50% of cases occur in association with MG. Thymoma occurring in association with MG, should always be surgically removed ( 17 ). Minimally invasive surgical approaches are feasible in most but may not be possible in the larger tumors. Complete surgical resection is aimed for but radiotherapy may be required for invasive thymomas. Where resection is incomplete and/or surgical margins are positive for thymoma, radiotherapy improves the prognosis by 50–60% ( 53 , 54 ). Thymomas are also chemosensitive. Platinum-based agents show consistent efficacy ( 55 ) and can improve the outcome of Masaoka stage III and IV thymomas or recurrent thymomas. Non-platinum based regimens are also prescribed in some tumors and the role of immunotherapy still remains to be further investigated.
Isolated ocular myasthenia is rare. While ptosis and diplopia are common presenting symptoms including in patients who will eventually evolve into generalized myasthenia, only 20% of patients will turn out to have pure ocular MG—signifying that these patients will never develop generalized disease) ( 56 , 57 ). The diagnostic difficulty with this entity is that only 50% have detectable antibodies to the AChR ( 57 ). Single fiber EMG studies support the diagnosis of neuromuscular transmission failure in patients without detectable antibodies, including ocular myasthenia ( 58 ). The main differential diagnoses include thyroid eye disease, and a progressive external ophthalmoplegia associated with a mitochondrial disorder. The latter group of patients may also have some minor abnormalities on single fiber EMG studies with borderline increased jitter values making the diagnosis even more challenging ( 58 ). Other diagnostic cues are therefore crucial, and ultimately a muscle biopsy may be necessary to clinch the diagnosis.
First-Line Pharmacological Therapy in Ocular MG
While ocular MG is not life-threatening, diplopia is a very disabling symptom. It significantly impacts an individual's quality of life—it impacts patients' driving ability, it may impact their employment, their social life, and their hobbies including sports, reading etc. When a patient presents with ocular myasthenia, the first treatment that should be initiated is pyridostigmine in order to achieve symptom control and to determine reversibility. This may be sufficient in patients with mild symptoms and signs, but is unlikely to be adequate in patients with significant ocular motility disturbance. If patients remain symptomatic despite maximal doses of pyridostigmine, then the next step should be prompt treatment with corticosteroids ( 59 – 63 ). Early treatment of ocular myasthenia improves the chances of reversibility or significant improvement in the long-term ( 64 ). There is some evidence to indicate that early treatment of ocular myasthenia delays or prevents the development of generalized disease ( 64 – 66 ). Delaying corticosteroid treatment, in our experience, reduces the chances of recovery of the extraocular muscles. In some patients, in spite of prompt and adequate treatment, they do not respond to therapies and are left with a fixed ophthalmoplegia in the absence of any other signs or symptoms. This may reflect the complex sarcomeric organization ( 67 , 68 ), gene expression ( 69 ), distinct complement expression ( 68 , 70 ), and unique metabolic demands and vulnerability of mitochondrial oxidation pathways within the extraocular muscles ( 71 ) that are susceptible to disease including autoimmune disorders. In patients who are refractory to treatment, and especially when they have no detectable antibodies and/or equivocal SFEMG findings, there is scope for investigating with an MRI scan of the orbits with gadolinium to exclude alternative, namely inflammatory, processes for instance thyroiditis. Commonly in ocular myasthenia patients with refractory disease and fixed ophthalmoplegia, the MRI shows atrophic extraocular muscles with asymmetric involvement and with no enhancement following gadolinium administration.
The ceiling steroid dose in ocular myasthenia is deemed to be lower than that used in generalized myasthenia ( 16 , 57 ). One usually aims for a maximal steroid dose (prednisolone/prednisone) of around 25 mg daily (or equivalent of 50 mg alternate days) but in some instances higher doses may need to be considered particularly if there is a delay in the correction of the ocular motility disturbance and if diplopia remains a persistent symptom. Recovery of the extraocular muscles in ocular myasthenia may take several months and there may be scope for adding in immunosuppressive agents for the same reasons as in generalized MG ( 16 , 22 , 72 ). The indications for this includes patients whose ocular motility does not respond to corticosteroids alone, or who experience frequent relapses and are unable to reduce the corticosteroid dose below an acceptable level, or the physician feels that additional treatment is required especially if there has been incomplete response to corticosteroids and pyridostigmine. Other patients are unable to tolerate corticosteroids or may have comorbidities such as diabetes, osteoporosis, depression, or glaucoma that preclude the long-term use of steroids.
It is important to monitor patient's response to treatment carefully and working with an orthoptist can be of immense assistance. There also needs to be an objective assessment of ptosis and ocular motility for instance using the Jampolsky scheme ( 73 , 74 ), and collaborative work with an orthoptist is often very helpful in monitoring response to treatment and progress.
Non-pharmacological Therapies in Ocular MG
In the short term, patients may be fitted with a Fresnel prism to allow some correction of their double vision ( 75 ). Reducing the strength of the prism over time is a clear indication of response and improvement. Some patients, however, will continue to rely on their prism in the long-term. Using a patch over one eye in the short-term is another option for some patients to help obliterate the false image while others tolerate using an occlusive contact lens.
Residual ptosis can be a significant problem in some patients either causing obstruction of one's vision or from an aesthetic perspective. In older patients, ptosis may be aggravated further by senile dehiscence or dermatochalasia. In general, if patients' ptosis does not reverse in spite of maximal treatment received over a 2-year period, then the chances of recovery after that period of time are rather slim. In a select group of patients, ptosis repair surgery performed by an oculoplastic surgeon may be indicated ( 72 , 76 ). The surgeon needs to ensure that the risk of corneal exposure is minimal and repeated procedures are best to be avoided. In contrast using ptosis props is a less invasive way of dealing with the problem but some patients complain that these cause discomfort or corneal dryness since the props limit blinking, and may be simply impractical for some. In some patients, the extraocular motility may remain abnormal in spite of adequate treatment with steroids, and may become fixed. In a highly select group, strabismus surgery may be of benefit but careful discussion with an ophthalmologist who specializes in squint surgery is required for these cases. Botulinum toxin to correct the strabismus should be avoided altogether in MG because of the toxin's systemic effects, which may destabilize MG patients even when their status (other than their ocular features) has been stable for many years ( 77 ).
Thymectomy in ocular myasthenia remains controversial but there are various small studies indicating that this is beneficial particularly when considered early in the disease ( 78 – 82 ). The task force for the EFNS/ENS guidelines ( 62 ) agreed that thymectomy is not recommended for ocular myasthenia as first-line treatment but should be considered if drug therapy was not successful and may prevent MG generalization (good practice point). Given that ocular myasthenia often evolves into generalized disease (and there are no markers to predict this) and given the increased access to minimally invasive thymectomy surgery, early intervention may be of benefit. For these reasons, we have increasingly been referring ocular MG patients for thymectomy in the last 3 years. Furthermore, it is unknown, if early thymectomy may also prevent these patients developing a fixed ophthalmoplegia in the long-term.
MG in Specific Patient Groups
The pregnant patient.
In practice, the majority of MG patients, who are treated adequately before pregnancy, do not experience any complications during pregnancy or in the post-partum phase. However, some report an increased risk of MG relapse during pregnancy that varies between 17% ( 83 ) to 41% ( 84 ). Some patients' MG status improves during pregnancy ( 85 ) as one observes with other autoimmune conditions such as multiple sclerosis. In the ideal scenario, the pregnancy is planned to allow optimization of MG status and withdrawal of teratogenic medications where relevant. Pyridostigmine, corticosteroids, and azathioprine are all safe to be used in pregnancy and should not be discontinued during pregnancy ( 85 , 86 ). MMF and Methotrexate are teratogenic and should be avoided ( 85 ). Ciclosporin and tacrolimus are not teratogenic but their use can be associated with the development of hypertension and gestational diabetes and, therefore, the patient requires close monitoring ( 85 ). IvIG and PE are also safe to be used during pregnancy ( 87 ). There are some reports suggesting that MG patients are at risk of preterm rupture of membranes ( 88 , 89 ). We have not encountered this in our practice, however.
Therapy for MG should be optimized where possible before and during pregnancy. The neurologist and obstetrician should be in regular dialogue particularly in the third trimester of pregnancy, when plans should be initiated on how the baby should be best delivered. Medications for MG should continue uninterrupted before and throughout labor. Patients should undergo spontaneous vaginal delivery in most cases and epidural labor analgesia should be considered early in patients who are likely to experience fatigue during labor ( 90 ). Nitrous oxide is safe to use ( 85 ).
Surgical delivery should be considered in those who MG status is poorly controlled and in those patients where muscle weakness is significant or their MG is considered brittle. Ideally this should be planned adequately in advance with multidisciplinary team discussions throughout but especially in the latter part of the pregnancy. MG patients are usually extremely sensitive to depolarizing muscle relaxants, and should be administered the least possible dose ( 85 ). Magnesium sulfate for the treatment of eclampsia should be avoided in MG since this will exacerbate myasthenic weakness ( 85 ). Opiates for pain relief especially in the post-partum phase should be used with caution since they too may exacerbate weakness. Breast feeding of the newborn should be encouraged. Neonatal myasthenia, with temporary and usually mild myasthenic weakness, occurs in 10% of neonates due to transplacental transfer of antibodies ( 86 , 91 ). It usually resolves spontaneously within 3 weeks of the birth of the infant. Rarely the presentation of the neonate can be more complex, especially if the mother's MG was undertreated during pregnancy, and may require the neonate to be managed in an intensive care setting for a short period. Very rarely, infants of MG mothers are born with mild myopathy and—at the severe end of the spectrum—arthrogryposis multiplex congenita ( 92 ). The mothers may in fact be asymptomatic or minimally symptomatic with elevated AChR antibodies and some may be asymptomatic with antibodies specific to the fetal AChR γ subunit ( 92 ).
The MG Patient in Crisis
MG crisis occurs in circa 20% of MG patients who are newly presenting with MG ( 93 , 94 ). It occurs more frequently in MG patients who are undertreated, or who have newly presented and whose treatment is being slowly escalated but whose presentation has evolved more rapidly than therapy has originally been scheduled for stabilization. Patients develop severe muscle weakness including weakness of the respiratory muscles, commonly preceded by severe bulbar weakness with dysphagia, with or without palatal weakness and nasal escape. In this situation, patients require a nasogastric tube to be inserted to allow medications to be administered and for feeding. This clinical picture must be promptly recognized and the patient requires to be monitored closely in hospital, usually in a high dependency unit setting, since this clinical picture often evolves further with significant respiratory muscle weakness. Arterial blood gases should be checked to identify when type 2 respiratory failure occurs. At the bedside, assessing the patient's respiratory rate and forced vital capacity, and observing whether the patient is using their accessory muscles are all helpful measurements, predictors or cues. If parameters allow then the patient could be treated with non-invasive ventilation (NIV) first but if parameters fail to improve or the patient continues to tire with NIV or is intolerant of this, then treatment must be quickly escalated and the patient must be intubated and mechanically ventilated in an intensive care setting.
The two primary pharmacological therapies to treat MG crisis are ivIG, at a dose of 0.4 g/kg/day for 5 days or PE—usually 4–6 exchanges ( 17 ). They are equally effective in the treatment of MG crisis or a significant MG relapse ( 95 ). We commonly prescribe ivIG first, unless there are contraindications, and resort to PE as second-line therapy if the patient fails to respond to ivIG. However, if PE is readily available we would recommend using as first-line in the context of MG crisis since it is more rapid in its effect than ivIG. This has been our experience and also previously shown by Qureshi et al. ( 96 ). PE is not without risk however. It is more invasive, more labor-intensive and more expensive than ivIG ( 97 ). PE should be performed via peripheral venous access, where feasible, but central catheters may be necessary in some which pose additional risks of an infection source if mishandled or if left in situ for too long ( 98 ). The same dose of ivIG could be administered over a shorter period for example 2–3 days if tolerated by the patient. We prefer to administer over 5 days, especially in patients who are ivIG naïve at least initially, and we consider administering over 2–3 days in subsequent treatments.
Corticosteroids are added or increased simultaneously with ivIG or PE therapy ( 16 ). In our practice, we still initiate corticosteroids at low doses but then we escalate the dose more rapidly over 5–7 days, since the steroid dip is likely to be counteracted by the simultaneous use of ivIG or PE. The role of acetylcholinesterase inhibitors is limited in MG crisis. They may exacerbate bronchial secretions and so one should be mindful of identifying the clinical situation when they are likely to be of benefit even to the MG patient in crisis. Some patients may require further courses of PE or ivIG 4–5 weeks after their initial therapy and may relapse even after their initial significant improvement. This is because the effect of corticosteroids may be apparent after 6–8 weeks while the effect of ivIG or PE usually lasts circa 4 weeks.
Weaning from the ventilator should be considered when the patient demonstrates an improvement in vital capacity and is strong enough to transition to spontaneous mode ventilation, which allows the patient to initiate breathing ( 99 ). The patient should be observed for fatigability with switch-over to assisted-ventilation when they fatigue. There is concomitant improvement in bulbar and neck muscle strength when respiratory muscle improvement is observed. If their cough remains weak and the patient is struggling to clear their airways secretions, then extubation is likely to be precocious and failure is more likely to occur.
Consideration for thymectomy should be considered where relevant and after the patient has been weaned off ventilation and extubated. Also, they should demonstrate stability in their MG status, have been stepped down to a regular ward and are becoming less dependent for their daily activities of daily living. The prognosis of MG crisis is worse in patients with thymoma. In this group of patients, managing their MG crisis can be challenging and response to therapy may be delayed ( 93 ). When their MG status has been stabilized, however, thymectomy should follow on promptly when safe to do so.
The Older MG Patient
World-wide epidemiological studies confirm that the incidence of MG is increasing among male and female patients who are older than 65 years ( 100 – 102 ) and the prevalence is also rising due to patients living longer ( 103 , 104 ). Multiple comorbidities often exist in older patients. They are less likely to tolerate the more potent immunosuppressive agents that benefit the younger MG patients ( 105 – 107 ). In older patients, careful consideration needs to be given of the potential impact of corticosteroid treatment on other systems for example the development of diabetes, hypertension, obesity with cardiac strain and heart failure, significant osteoporosis with vulnerability to various fractures. They become more vulnerable to infection including recurrent infections and sometimes resulting in life-threatening sepsis especially when more potent immunosuppressive agents such as MMF or Methotrexate are prescribed. Some older patients suffer recurrent infections when managed with maximal immunosuppression for their MG which in turn results in hospitalization, further deconditioning and a significant delay in recovery from their MG. From our experience, we have noted that in the older perhaps frailer patients it may be safer in the longer term to slightly undertreat their MG rather than aim to induce remission, since prescribing conventional doses of immunosuppression in this age-group often leads to fatal consequences. MG patients are also more vulnerable to developing osteoporosis ( 108 ) and the prescribing neurologist needs to be aware of this and monitor closely patients' bone densities since osteoporotic fractures result in significant morbidity, chronic pain and reduced mobility, which may already be compromised in an older patient.
The Refractory MG Patient and Novel Therapies
About 20% of MG patients are refractory to all conventional treatments. Monoclonal antibody treatments that bind the B lymphocyte membrane protein CD20, such as Rituximab have been increasingly prescribed in this group of patients with successful outcomes. The rationale behind preparations such as Rituximab is that they destroy and deplete pathogenic B cells and decrease AChR antibody production. Rituximab influences the whole spectrum of B cell function including antigen presentation, cytokine production, and T cell stimulation and hence has a role in T cell mediated autoimmune diseases too ( 109 ). Studies have demonstrated that clinical improvement even with one cycle of Rituximab is sustained ( 110 , 111 ) allowing subsequent reduction in steroid doses and in some inducing remission ( 112 ). Patients with MuSK-MG respond extremely well to Rituximab and the drug often induces remission without the requirement for subsequent infusions ( 112 , 113 ). Rituximab has a role in patients presenting aggressively and explosively at onset and who are refractory to all conventional therapies. Brauner et al. ( 114 ) demonstrated that clinical outcomes were better in patients who were treated early rather than later with Rituximab. There is scope for considering Rituximab in patients who are in crisis and who are not responding to high dose corticosteroids or ivIG or PE, and when patients demonstrate resistance in weaning off ventilation during the treatment pathway of MG crisis. Caution must be exerted in this scenario, acknowledging that Rituximab will not be effective immediately and may pose an added risk to the patient for developing infection. Rituximab is contraindicated during pregnancy ( 87 ).
In our experience, where we have treated a small cohort of 17 MG patients with MuSK-MG, AChR-MG, and MG with no detectable antibodies, the majority of patients improved significantly but remain dependent on immunosuppression (unpublished data). Our single MuSK-MG patient, within this small cohort, responded best to Rituximab although this did not induce complete remission of her disease. In contrast, about a third of MG patients did not respond to Rituximab and their MG status was not altered by this therapy. In general, we have found that the drug is well-tolerated with minimal side effects. However, in two patients we have observed delayed neutropenia developing many months after Rituximab treatment, including one patient whose presentation was complicated by two neutropenic sepsis episodes several months after their Rituximab treatment. This has been observed in other patient groups treated with Rituximab ( 115 – 117 ).
In a large systemic review of 169 MG patients who received Rituximab, remission (PR or CSR) and MM was achieved in 72% of MuSK-MG patients in contrast to 30% of AChR-MG patients, with post-treatment relapses being markedly reduced in the MuSK-MG cohort ( 118 ). It is still unclear why MuSK-MG patients respond so well to this drug. It would be crucial for biomarkers to be developed that will allow physicians to predict a patient's response to Rituximab. There is also a similar crucial need for robust trial data for this drug, since the efficacy of Rituximab in AChR-MG is still debatable and the studies that are available may be limited by an element of reporting bias ( 119 ). This data will also help physicians counsel patients adequately when embarking on this therapy.
MG treatment can also be addressed by switching off complement pathways and their activation, or by altering the Fc region of the antibody such that less antibodies are available for recycling, more are destroyed and thus unavailable for pathogenic processes. Novel therapies have been developed to address both. The efficacy and safety of the terminal complement inhibitor eculizumab (a humanized monoclonal anti-C5 antibody) in MG has been rigorously studied in the REGAIN trial ( 120 , 121 ). Improvements were noted in all objective MG-related scores and in the patients' quality of life scores for all those actively treated with eculizumab, and were sustained during the 52-week study period. Patients treated in the placebo arm experienced rapid and sustained improvement in their MG status when switched to open-label eculizumab. The drug also improved fatigue scores which in turn correlated strongly with MG-specific outcome measures ( 122 ). However, the response among patients in the REGAIN trial was variable with some improving substantially, some modestly and some patients showing no response whatsoever ( 123 ). Eculizumab is now a registered therapy for myasthenia gravis. It remains an expensive drug with costs for one patient's treatment per annum amounting to $500,000. It is unclear whether this drug is cost-effective in MG. A trial of zilucoplan, a subcutaneously self-administered inhibitor of complement component 5, has been recently studied ( 124 ). The trial confirmed that zilucoplan was safe and well-tolerated and patients rapidly showed clinical improvement with this drug. The extent of clinical response correlated with the level of complement inhibition such that near-complete inhibition was demonstrated to be superior to submaximal inhibition.
Efgartigimod (also known as ARGX-113) has been trialed in generalized MG in a phase-2 randomized double-blind, placebo-controlled study in 15 centers ( 125 ). ARGX-113 is the anti-neonatal Fc receptor immunoglobulin IgG1 fragment. It has been modified to increase its normal affinity for IgGs, thus blocking the formation of disease-causing IgG. Efgartigimod was well-tolerated in this trial. In the 12 patients treated with the active drug, there was a rapid decline in total Ig levels and in AChR titers, which in turn correlated with a clinical improvement of their MG, and this was sustained in the majority.
The proteasome inhibitor, Bortezomib, depletes short-lived and long-lived B cells and is applied in the treatment of multiple myeloma ( 126 ) and plasmablastic lymphoma ( 127 ). It is likely to have a role in the treatment of refractory MG including MuSK antibody positive MG ( 128 ) but the development of a sensorimotor polyneuropathy, a recognized side-effect of this drug, is likely to be a limiting factor.
Questions remain unanswered about the long-term safety, efficacy, and tolerability of these novel therapies (meaning after several years of continuous treatment). It is unclear whether long-term complement inhibition, for instance, would pose increased general infection risks particularly in older age-groups. Determining the category of patients who are likely to benefit from these therapies is crucial. Would these therapies be aimed only for “refractory” and “severe” MG? If so, how do we precisely define these entities? Would drug holidays be considered and if so for how long? It is also less clear how cost-effective these novel therapies are, how the various global health systems would fund these drugs and how the different health insurance companies will cover the costs of these drugs. A detailed cost-utility analysis is required that will allow the diverse health systems to better understand the long-term efficacy of these therapies, how improvements in objective measurements translate into better function for the patient, and how they improve patients' quality of life. It would be imperative to ascertain and quantify the potential socioeconomic gains when using these therapies (do these therapies allow individuals to return to their employment, increase independence and reduce dependence on care-givers?) and the impact on reducing in-patient hospital care (reducing hospital admissions including to intensive care units, the requirement for regular ivIG, or frequency of attend clinic appointments due to stable disease etc.).
Fatigue in MG
Fatigue is common in all neuromuscular conditions including MG, and around 80% of MG patients will experience significant fatigue at some stage of their disease ( 129 , 130 ). It is distinct from fatigability and muscle weakness and therefore it is crucial that the physician recognizes this entity since its management does not involve escalation of treatment for MG ( 131 ). Fatigue is as disabling to the patient as active muscle weakness, and may negatively impact patients' quality of life, their quality time with their family, their employment status, and their social lives. It contributes to the disease burden but is more difficult to assess or objectively measure in the clinic. Fatigue may be problematic even when MG symptoms have largely settled or when the patient has achieved minimal manifestations.
Fatigue is multifactorial. Primary fatigue occurs when muscle weakness and fatigability are active in MG and has an inherent physical component ( 132 ) contributing to fatigue. They also complain of cognitive fatigue which patients often allude to as “brain fog” ( 133 ). It is difficult to dissect out primary from secondary fatigue, with the latter occurring for various reasons. Patients with MG, often gain weight primarily due to corticosteroid treatment ( 134 ), sleep less efficiently ( 135 ), move less and develop muscle stiffness and discomfort ( 136 ). They are more likely to become anxious and depressed about their physical limitations and the variability and unpredictability of their symptoms ( 137 ). They resort to socializing less, they might discontinue their employment, which in turn may have financial consequences, and do less chores in the house or even become virtually house-bound. O'Connor et al. ( 138 ) identified that MG patients were more likely to become sedentary even when asymptomatic. It is unclear whether this is learnt behavior or fatigue-driven or simply part of a vicious cycle. Because MG patients exercise less they become quickly deconditioned and often develop breathlessness that is not secondary to respiratory muscle weakness. Their breathing becomes shallow with a tendency to hyperventilate which develops as a learned pattern and is often misinterpreted as a sign of early MG crisis. Their sleep pattern is less efficient. They may develop obstructive sleep apnoea due to weight gain. They socialize less and this in turn negatively impacts their mood further.
Fatigue is not unique to MG but is also prevalent in other neuromuscular disorders such as different types of muscular dystrophy and myotonic dystrophy (DM1). Patients with facioscapulohumeral muscular dystrophy (FSHD) often complain of fatigue and pain, and hypersomnolence is very common in patients with myotonic dystrophy. Various studies have studied the role of exercise in various neuromuscular studies including MG ( 139 , 140 ). Other studies have explored using cognitive behavioral therapy in combination with graded exercise in MG, DM1, and FSHD including high intensity training and aerobic exercise which led to functional benefits in patients without evidence of damaging muscle ( 141 – 144 ). In a very small and select group of patients, where fatigue is compounded by pain, anxiety and insomnia, and perhaps with an overlay of their myasthenic symptoms (i.e., true MG coexisting with an aspect of a functional neurological disorder) we have managed them also with psychology input and cognitive behavioral therapy ( 145 ).
It is challenging when prescribing exercise to MG patients or indeed to any neuromuscular patient. Different types of exercise are suitable for MG patients at different phases of their MG. Aerobic or high intensity training is not possible when MG patients are very symptomatic. In this situation, stretching exercises such as Tai chi, slow flow yoga or pilates are probably most appropriate with emphasis also on balance maintenance. When MG symptoms stabilize, physical therapy should focus on balance and muscle strengthening but physicians should also enquire specifically about other symptoms including pain, residual fatigue, sleep disturbance and mood problems and address these accordingly.
Dysfunctional Breathing in Myasthenia Gravis
It has long been observed that breathing patterns and the central ventilator drive can be altered in patients with mild or moderate MG ( 146 ). In our practice, we have observed several patients, who we deem stable or in minimal manifestations, complaining of dyspnoea as a residual prominent symptom in spite of them not having any objective evidence of respiratory muscle weakness. A very small proportion, may have had a MG crisis at some stage of their disease, which inevitably raises long-term anxiety levels to the patient and their carer, about the potential severity and sometimes unpredictability of the disease. In some, contributory factors are clear and include deconditioning or weight gain. We have identified, through collaborative work with the local respiratory team, that many of these patients have developed dysfunctional breathing (unpublished observation). Our local respiratory physiotherapist has been working with these patients, employing physiotherapy-based breathing pattern modification interventions. These include relaxation of intercostal muscles, accessory muscles and full utilization of the diaphragm thus helping them to regulate and improve their breathing pattern with good results (unpublished).
Dysfunctional breathing has been studied extensively in poorly controlled asthma ( 147 ) because it is common and is associated with significantly poor asthma control and lower quality of life. Evidence-based guidelines recommend breathing retraining interventions as adjuvant treatment in uncontrolled asthma. A multicenter randomized controlled trial is currently underway in Denmark to investigate the effect of breathing retraining on the impact on quality of life in poorly controlled asthmatics ( 148 ). In a small study ( 149 ), 12 MG patients underwent long-term respiratory muscle endurance training, which resulted in a change in their breathing pattern with prolonged expiration. Interestingly patients reported an improvement in their MG symptoms, in their respiratory symptoms and in their physical fitness. This study proves that normocapnic hyperpnea training is a useful adjuvant therapy in MG.
It is therefore imperative that physicians recognize the entity of dysfunctional breathing in MG patients and refer them on for respiratory-based physiotherapy. This is a crucial adjuvant treatment in MG patients, who complain of dyspnea, and intervention helps their overall MG symptoms, improves their exercise capacity and increases their chances of overall recovery with improved quality of life.
The End-Result—Our Practice and Comparison With Reported Outcomes
When we set up the myasthenia clinic 13 years ago, we primarily aimed this to be a regional service that manages MG patients residing in the West of Scotland. However, we were subsequently referred MG patients who were refractory to standard therapies and who came from other parts of Scotland. Our patient cohort, served over a 13-year period, is heterogeneous including ocular and generalized MG, spanning all age groups (including patients in their tenth decade), with different antibody status and thymic pathology. About 10% of our cohort is refractory to conventional treatments. Our experience, as previously reported in the literature ( 150 ), has been that most patients' MG status evolves within the first 2 years of symptom onset. Broadly, CSR has been achieved in 5–10% of our case-load, PR in 20%, MM in 25%, improvement in 35%. About 10% of our cohort's MG status remains unchanged by our therapeutic interventions. Patients were worsened by therapy in 1–2%, and 1% died from direct complications of their MG. Our rate of PR is comparable to what has been reported in the literature but it is difficult to make direct comparisons since our treatment regime has also evolved over time. Mantegazza et al. ( 151 ) reported PR in 24% and CSR in 11%. Beghi et al. ( 152 ) reported a higher chance of CSR in patients who were younger and who had a shorter disease duration. These findings were echoed in a further study by the same group almost a decade later ( 153 ). Yang et al. ( 154 ) reported a CSR rate of 60% in patients who received thymectomy for thymic hyperplasia with younger patients having a higher CSR rate. Given that we have put more MG patients forward for thymectomy in the last 3–4 years, it is likely that this would further influence our remission rates. If we were to categorize our patient cohort according to age-groups, thymus pathology, and thymectomy status this would refine our CSR and PR rates, but we have not carried out that detailed analysis to date.
Conclusions
There are various guidelines in the literature on MG management. Physicians usually adhere to and achieve confidence and familiarity with specific treatment plans. However, the “recipe” for treatment can and should be designed for the individual patient's comorbidities. The aim in MG treatment is to induce remission or MM and to enable patients to resume their normal life-style. Each patient, however, is unique with respect to their comorbidities and their social or personal circumstances. As a result, the immunosuppressive therapy prescribed needs to be “catered” for that particular individual bearing all those pertinent variables in mind. Residual myasthenic symptoms, which physicians may perceive as minimal may have a significant impact on a patient's daily life. As physicians, we need to be mindful of the impact of patients' MG on their physical and mental health, the impact on their family or carers, and the impact of adverse effects from MG-related therapies on their general health. The development of new therapies for the severe end of the MG spectrum is exciting. We need to learn more about these drugs, gain familiarity and identify the patient groups who are more likely to benefit from them. Detailed cost-utility analysis is required for individual health-care systems to enable physicians in their process of justifying the use of these drugs to their respective hospital systems. Addressing fatigue and its management is paramount to the overall MG management. Encouraging patients to exercise should be an integral part of their treatment since this will help their overall well-being in the long-term. Finally, dysfunctional breathing should be recognized and treated accordingly.
Author Contributions
MF and JG contributed equally to conceptualizing this document. MF wrote the manuscript with contributions from JG to different sections. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Ms. Caroline Carmichael, myasthenia nurse specialist, who makes our busy service possible and sustainable.
Abbreviations
Real-World experience with efgartigimod in patients with myasthenia gravis
- Original Communication
- Published: 25 March 2024
Cite this article
- Lior Fuchs 1 ,
- Shahar Shelly 2 , 3 ,
- Ifat Vigiser 1 , 4 ,
- Hadar Kolb 4 ,
- Keren Regev 4 ,
- Yoel Schwartzmann 5 ,
- Adi Vaknin-Dembinsky 5 , 6 ,
- Amir Dori 1 , 7 &
- Arnon Karni ORCID: orcid.org/0000-0002-5625-7900 1 , 4 , 8 , 9
87 Accesses
Explore all metrics
Recommendations for the treatment of myasthenia gravis (MG) have been difficult to develop because of limited evidence from large randomized controlled trials. New drugs and treatment approaches have recently been shown to be effective in phase 3 studies in seropositive generalized (g) MG. One such drug is efgartigimod, a human-Fc-fragment of IgG1, with a high affinity for the endosomal FcRn. We conducted a multicenter study to evaluate the real-world clinical and safety effects of efgartigimod in 22 gMG patients. We evaluated the strategies for the timing of re-treatment with it. The participants received a total of 59 efgartigimod -treatment cycles. The median number of cycles was 2 (range 1–6). Twenty patients (86.3%) improved by at least 2 MG-ADL points after the first treatment cycle. The median MG-ADL score at baseline was 6.5 (range: 3–17) and 2.5 (range: 0–9) post-treatment ( p < 0.001). A consistent improvement of at least 2 points in the MG-ADL score after each cycle occurs in 18 patients. The effect duration of the treatment was usually between 4 and 12 weeks. Two major clinical patterns of treatment response were found. Treatment with efgartigimod was also associated with significant reductions of prednisone doses Overall, the treatment was safe and associated with only minor adverse events. The single fatality was apparently due tosevere respiratory failure. We found that efgartigimod is clinically effective, may be used as a steroid sparing agent and is generally safe for gMG patients. We recommend a personalized preventive treatment approach until clinical stabilization, followed by discontinuation and periodic evaluations.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
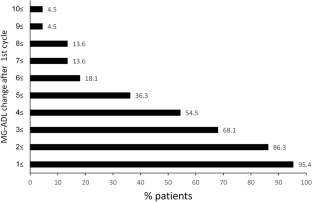
Similar content being viewed by others

Drugs Targeting CD20 in Multiple Sclerosis: Pharmacology, Efficacy, Safety, and Tolerability
Alise K. Carlson, Moein Amin & Jeffrey A. Cohen

Long-term Efficacy and Safety Following Switch Between Upadacitinib and Adalimumab in Patients with Rheumatoid Arthritis: 5-Year Data from SELECT-COMPARE
Roy Fleischmann, Ricardo Blanco, … Eduardo Mysler

Systemic Lupus Erythematosus (SLE) Therapy: The Old and the New
Fabio Basta, Federica Fasola, … Andreas Schwarting
Data availability
The research data is available and will be provided after contacting the corresponding author.
Vincent A, Newsom-Davis J (1985) Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J Neurol Neurosurg Psychiatry 48(12):1246–1252
Article CAS PubMed PubMed Central Google Scholar
Grob D, Brunner N, Namba T, Pagala M (2008) Lifetime course of myasthenia gravis. Muscle Nerve 37(2):141–149
Article PubMed Google Scholar
Drachman DB (1994) Myasthenia gravis. N Engl J Med 330(25):1797–1810
Article CAS PubMed Google Scholar
Berrih-Aknin S, Le Panse R (2014) Myasthenia gravis: a comprehensive review of immune dysregulation and etiological mechanisms. J Autoimmun 52:90–100
Dresser L, Wlodarski R, Rezania K, Soliven B (2021) Myasthenia gravis: epidemiology, pathophysiology and clinical manifestations. J Clin Med 10(11):2235
McGrogan A, Sneddon S, de Vries CS (2010) The incidence of myasthenia gravis: a systematic literature review. Neuroepidemiology 34(3):171–183
Skeie GO, Apostolski S, Evoli A, Gilhus NE, Illa I, Harms L, Hilton-Jones D, Melms A, Verschuuren J, Horge HW (2010) Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol 17(7):893–902
Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, Kuntz N, Massey JM, Melms A, Murai H, Nicolle M, Palace J, Richman DP, Verschuuren J, Narayanaswami P (2016) International consensus guidance for management of myasthenia gravis: executive summary. Neurology 87(4):419–425
Article PubMed PubMed Central Google Scholar
Narayanaswami P, Sanders DB, Wolfe G, Benatar M, Cea G, Evoli A, Gilhus NE, Illa I, Kuntz NL, Massey J, Melms A, Murai H, Nicolle M, Palace J, Richman D, Verschuuren J (2021) International consensus guidance for management of myasthenia gravis: 2020 update. Neurology 96(3):114–122
Gilhus NE (2016) Myasthenia gravis. N Engl J Med 375(26):2570–2581
Jani-Acsadi A, Lisak RP (2010) Myasthenia gravis. Curr Treat Options Neurol 12(3):231–243
Gilhus NE, Verschuuren JJ (2015) Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol 14(10):1023–1036
Pascuzzi RM, Coslett HB, Johns TR (1984) Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 15(3):291–298
Howard JF Jr, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, Jacob S, Vissing J, Burns TM, Kissel JT, Muppidi S, Nowak RJ, O’Brien F, Wang JJ, Mantegazza R (2017) Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol 16(12):976–986
Meisel A, Annane D, Vu T, Mantegazza R, Katsuno M, Aguzzi R, Frick G, Gault L, Howard JF (2023) Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: results from the phase 3 CHAMPION MG open-label extension. J Neurol 270(8):3862–3875
Howard JF Jr, Bresch S, Genge A, Hewamadduma C, Hinton J, Hussain Y, Juntas-Morales R, Kaminski HJ, Maniaol A, Mantegazza R, Masuda M, Sivakumar K, Śmiłowski M, Utsugisawa K, Vu T, Weiss MD, Zajda M, Boroojerdi B, Brock M, de la Borderie G, Duda PW, Lowcock R, Vanderkelen M, Leite MI (2023) Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol 22(5):395–406
Howard JF Jr, Bril V, Vu T, Karam C, Peric S, Margania T, Murai H, Bilinska M, Shakarishvili R, Smilowski M, Guglietta A, Ulrichts P, Vangeneugden T, Utsugisawa K, Verschuuren J, Mantegazza R (2021) Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 20(7):526–536
Bril V, Drużdż A, Grosskreutz J, Habib AA, Mantegazza R, Sacconi S, Utsugisawa K, Vissing J, Vu T, Boehnlein M, Bozorg A, Gayfieva M, Greve B, Woltering F, Kaminski HJ (2023) Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol 22(5):383–394
Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS (2019) The neonatal Fc receptor (FcRn): a misnomer? Front Immunol 10:1540
Ulrichts P, Guglietta A, Dreier T, van Bragt T, Hanssens V, Hofman E, Vankerckhoven B, Verheesen P, Ongenae N, Lykhopiy V, Enriquez FJ, Cho J, Ober RJ, Ward ES, de Haard H, Leupin N (2018) Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest 128(10):4372–4386
Howard JF Jr, Bril V, Burns TM, Mantegazza R, Bilinska M, Szczudlik A, Beydoun S, Garrido FJRR, Piehl F, Rottoli M, Van Damme P, Vu T, Evoli A, Freimer M, Mozaffar T, Ward ES, Dreier T, Ulrichts P, Verschueren K, Guglietta A, de Haard H, Leupin N, Verschuuren JJGM (2019) Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 92(23):e2661–e2673
Riley TR, Douglas JS, Wang C, Bowser KM (2023) An update of the pharmacological treatment options for generalized myasthenia gravis in adults with anti-acetylcholine receptor antibodies. Am J Health Syst Pharm 80(11):652–662
Wolfe GI, Ward ES, de Haard H, Ulrichts P, Mozaffar T, Pasnoor M, Vidarsson G (2021) IgG regulation through FcRn blocking: A novel mechanism for the treatment of myasthenia gravis. J Neurol Sci 430:118074
Heo YA (2022) Efgartigimod: first approval. Drugs 82(3):341–348
VYVGART 2021 (efgartigimod alfa-fcab) injection, for intravenous use. US Food and Drug Administration (FDA) approved product information. Revised December 2021. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761195s000lbl.pdf (Accessed on June 22, 2023).
Nowak RJ, Coffey CS, Goldstein JM, Dimachkie MM, Benatar M, Kissel JT, Wolfe GI, Burns TM, Freimer ML, Nations S, Granit V, Smith AG, Richman DP, Ciafaloni E, Al-Lozi MT, Sams LA, Quan D, Ubogu E, Pearson B, Sharma A, Yankey JW, Uribe L, Shy M, Amato AA, Conwit R, O’Connor KC, Hafler DA, Cudkowicz ME, Barohn RJ (2022) Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: the BeatMG study. Neurology 98(4):e376–e389
Lascano AM, Lalive PH (2021) Update in immunosuppressive therapy of myasthenia gravis. Autoimmun Rev 20(1):102712
Download references
No funding was received for conducting this study.
Author information
Authors and affiliations.
Faculty of Medicine & Health Sciences, Tel Aviv University, Tel Aviv-Yafo, Israel
Lior Fuchs, Ifat Vigiser, Amir Dori & Arnon Karni
Department of Neurology, Rambam Medical Center, Haifa, Israel
Shahar Shelly
Rappaport Faculty of Medicine, Technion-Israel Institute of Technology, Haifa, Israel
Neuroimmunology and MS Unit, Neurology Institute, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel
Ifat Vigiser, Hadar Kolb, Keren Regev & Arnon Karni
Department of Neurology, Hadassah-Hebrew University Medical Center, Jerusalem, Israel
Yoel Schwartzmann & Adi Vaknin-Dembinsky
Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem, Israel
Adi Vaknin-Dembinsky
Department of Neurology, Sheba Medical Center, Ramat-Gan, Israel
Sagol School of Neuroscience, Tel Aviv University, Tel Aviv, Israel
Arnon Karni
The Neurology Institute, Tel Aviv Sourasky Medical Center, 6 Weizmann Street, 6423906, Tel Aviv, Israel
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Arnon Karni .
Ethics declarations
Conflict of interest.
All the authors have no conflict interest to declare that are relevant to the content of this article.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Fuchs, L., Shelly, S., Vigiser, I. et al. Real-World experience with efgartigimod in patients with myasthenia gravis. J Neurol (2024). https://doi.org/10.1007/s00415-024-12293-5
Download citation
Received : 29 January 2024
Revised : 28 February 2024
Accepted : 29 February 2024
Published : 25 March 2024
DOI : https://doi.org/10.1007/s00415-024-12293-5
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Generalized myasthenia gravis
- Efgartigimod
- Find a journal
- Publish with us
- Track your research
Log in using your username and password
- Search More Search for this keyword Advanced search
- Latest content
- Current issue
- BMJ Journals More You are viewing from: Google Indexer
You are here
- Online First
- Epidemiology of myasthenia gravis in Denmark, Finland and Sweden: a population-based observational study
- Article Text
- Article info
- Citation Tools
- Rapid Responses
- Article metrics
- John Vissing 1 ,
- Sari Atula 2 ,
- Mari Savolainen 3 ,
- Juha Mehtälä 4 ,
- Laila Mehkri 5 ,
- http://orcid.org/0000-0002-6295-7399 Tina Bech Olesen 5 ,
- Tero Ylisaukko-oja 4 ,
- Ingrid Lindberg-Schager 6 ,
- Fredrik Berggren 7 ,
- http://orcid.org/0000-0001-8329-5219 Fredrik Piehl 8 , 9
- 1 Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet , University of Copenhagen , Copenhagen , Denmark
- 2 Clinical Neurosciences, Neurology , University of Helsinki and Helsinki University Hospital , Helsinki , Finland
- 3 UCB Pharma , Espoo , Finland
- 4 MedEngine Oy , Helsinki , Finland
- 5 MedEngine DK ApS , Copenhagen , Denmark
- 6 UCB Pharma , Stockholm , Sweden
- 7 UCB Pharma , Copenhagen , Denmark
- 8 Department of Clinical Neuroscience , Karolinska Institutet , Stockholm , Sweden
- 9 Department of Neurology , Karolinska University Hospital , Stockholm , Sweden
- Correspondence to Professor John Vissing, Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, 2100, Denmark; john.vissing{at}regionh.dk
Background Incidence and prevalence rates of myasthenia gravis (MG) vary considerably across studies, and mortality risk is rarely addressed. We examined the prevalence and incidence rates, mortality and factors associated with mortality with MG.
Method This was a registry linkage study based on nationwide health and administrative registries of Denmark, Finland and Sweden (populations of 5.9, 5.6 and 10.5 million, respectively). Patients with MG were identified based on International Classification of Diseases codes from inpatient and outpatient specialised care registries. Yearly prevalence, incidence and mortality rates in relation to the total background population were calculated from 2000 to 2020 (study period). The causes of death and factors associated with mortality were addressed separately.
Results The overall incidence of MG was 1.34 (95% CI 1.27 to 1.41), 1.68 (95% CI 1.60 to 1.75) and 1.62 (95% CI 1.56 to 1.68) per 100 000, and the overall prevalence per 100 000 was 18.56 (95% CI 18.31 to 18.81), 20.89 (95% CI 20.62 to 21.16) and 23.42 (95% CI 23.21 to 23.64) in Denmark, Finland and Sweden, respectively. The overall standardised mortality ratio (SMR) was 1.32 (95% CI 1.23 to 1.42) among patients with MG in Denmark, 1.23 (95% CI 1.15 to 1.33) in Finland, and 1.20 (95% CI 1.14 to 1.26) in Sweden, with higher SMR observed in women than men. Annual incidence and prevalence increased over time, whereas the SMR remained stable. The most common causes of death were MG, chronic ischaemic heart disease and acute myocardial infarction.
Conclusions This population-based study from three Nordic countries highlights the need for improved care of patients with MG, especially young women.
- EPIDEMIOLOGY
- CLINICAL NEUROLOGY
Data availability statement
Data may be obtained from a third party and are not publicly available. This is a registry study. Data may be obtained from a third party and are not publicly available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/ .
https://doi.org/10.1136/jnnp-2023-333097
Statistics from Altmetric.com
Request permissions.
If you wish to reuse any or all of this article please use the link below which will take you to the Copyright Clearance Center’s RightsLink service. You will be able to get a quick price and instant permission to reuse the content in many different ways.
WHAT IS ALREADY KNOWN ON THIS TOPIC
Existing literature reports a wide range in prevalence and incidence rates of myasthenia gravis (MG), but there are limited nationwide population-based epidemiological data available.
WHAT THIS STUDY ADDS
The incidence and prevalence of MG increased over time and a higher standardised mortality ratio was observed in patients with MG, especially in women. The study also describes causes of death and factors associated with the risk of death among patients with MG.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These findings are relevant for planning of healthcare resources and highlight an unmet medical need for improved therapeutic management of MG to reduce morbidity and mortality.
Introduction
Myasthenia gravis (MG) is a rare, chronic, autoimmune disease characterised by variable degrees of abnormal muscle fatigue in ocular, bulbar, axial and extremity skeletal muscle groups, sometimes leading to life-threatening respiratory insufficiency. 1 Symptoms of MG are caused by autoantibodies recognising receptors of the postsynaptic neuromuscular junction, interfering with their function and leading to deteriorated synaptic signalling and, subsequently, strength of skeletal muscles. 2 The acetylcholine receptor is by far the most common target (approximately 85% of patients), 3 with smaller proportions of patients being positive for autoantibodies for the muscle-specific kinase protein, while 10%–15% of patients are seronegative for any of the known target proteins. 1 4
MG is diagnosed based on clinical findings, presence of autoantibodies, neurophysiological tests showing disturbed neuromuscular transmission and treatment response to cholinesterase inhibitors, which increase the concentration of neurotransmitter acetylcholine at the neuromuscular junction. 1 Patients with MG can be further subgrouped based on the age of onset, antibody status, presence of thymus pathology and clinical presentation (ie, ocular myasthenia vs generalised myasthenia), all of which impact prognosis and treatment strategy. 4–6
In epidemiological studies of MG, the incidence rate has varied with age, sex and ethnic group. 7 However, differences in study population composition and criteria for case definitions likely partly explain the substantial variability among previous studies. Hence, the reported incidence ranges from 0.4 (Norway and Denmark) to as high as 2.9 (Sweden) and 4.6 (Germany) per 100 000. 8–11 Similarly, a review of 24 studies from 1990 to 2014 found that MG prevalence ranged from 5.4 to 35.0 per 100 000. 8 Only a few comprised larger population-based materials, but nationwide studies conducted in Sweden and Finland reported MG prevalence as 24.8 and 29 per 100 000, respectively. 12 13 Similar to other autoimmune conditions, the incidence and prevalence of MG reportedly increase over time, though it is unclear to what degree this is due to improved awareness and detection. 14
Improved therapeutic management has contributed to increased life expectancy in MG. 15 Nonetheless, mortality remains elevated in patients with MG. An overall mortality rate ratio of 1.41 with higher mortality among women was reported in Denmark 16 and a mortality ratio of 1.28 in patients with MG compared with the general population was reported in Sweden. 17
Collectively, the literature does not provide consistent information on the epidemiology of MG in contemporary populations over time, and few studies have examined, for example, factors associated with mortality. The Nordic countries, with universal access to publicly funded healthcare and high-quality health registries, offer excellent opportunities for population-based studies exploring the lifetime MG disease course. However, no study to date has leveraged data from more than one country. The objective of this study was to establish and cross-compare MG incidence, prevalence and mortality rates in Denmark, Finland and Sweden. In addition, causes of death and factors associated with the risk of death were examined.
Study setting and data sources
This was an observational, population-based cohort study using data from the nationwide health and administrative registries of Denmark, Finland and Sweden, with total populations of 5.9, 5.6 and 10.5 million, respectively. 18–20 All three countries have a tax-funded healthcare system with universal access to healthcare for all citizens, as well as digital population, health and social care registries with complete population-wide coverage. The standard of care of MG has largely followed international consensus guidelines in all three countries during the last two decades. 21
The identification of patients with MG and the comorbidity data was based on the national patient registries (The Danish National Patient Register, The Finnish Care Register for Health Care, and The Swedish National Patient Registry). Basic demographic information and data on migration were collected from The Danish Civil Registration System, Statistics Finland and The Swedish Population Registry. Cause of death information was collected from the causes of death registries of each country. In Finland and Sweden, the cause of death registries also includes information on the time of death, while in Denmark this information was collected from the Civil Registration System. Data from each registry can be extracted and linked through a unique personal identification number assigned to all individuals at birth or immigration, allowing follow-up of all study participants until death, emigration or end of data collection. In addition, all Nordic countries have central statistical agencies collecting and providing valid historical demographic data which served as the source of information on background population size and composition for epidemiological analyses. The diagnosis information was based on the International Classification of Diseases (ICD) codes.
Study cohorts
All patients with at least one diagnostic code for MG (ICD-10 codes G70.0*, ICD-9 codes 358.0*, ICD-8 code 73 309 and/or ICD-7 code 74400) in the national patient registries between 1 January 2000 (start of the study period) and 31 December 2020 (end of the study period) were included in the study.
Three cohorts were formed for the analyses. The full primary cohort (FPC) included all patients with at least one MG diagnosis and the prevalent subcohort (PSC) included all patients with at least two MG diagnoses in the study period. The incident subcohort (ISC) included all patients with at least two MG diagnoses during the study period, and no MG diagnoses before 1 January 2000. A minimum of 12 months without an MG diagnosis before the date of diagnosis was required for a patient to be included in the incident cohort (prior MG diagnosis was screened from all available registry data, starting from the start of the patient register in each country; year 1977 in Denmark, 1969 in Finland, and 1964 in Sweden). The last incident cohort date was 2019 in Finland and Sweden and 2020 in Denmark, due to the availability of data. The analyses of prevalence and incidence required at least two diagnoses of MG (initial and confirmatory) and were therefore based on the PSC and ISC. The mortality analysis was based on the PSC and was compared with the FPC. For all cohorts, the date of the first inpatient or outpatient specialist visit with an MG diagnosis was designated as the patient’s index date (MG diagnosis date). The baseline period was 12 months prior to the index date.
Statistical analyses
Descriptive statistics were used to calculate numbers and proportions for categorical variables; and means, medians, SD and first and third quartiles (Q1, Q3) for continuous variables. The incidence of MG was estimated in the ISC as the number of incident cases divided by the size of the total population in each country and expressed as the number of incident cases per 100 000 persons. The prevalence of MG was estimated in the PSC as of 31 December (the prevalence date) for each calendar year across the study period (2000–2020). Patients alive on the prevalence date and with the date of MG diagnosis any time before the prevalence date were included in the numerator, and the total background population alive on the prevalence date in the denominator. The expected annual number of deaths was estimated from the sex, age and calendar year-specific national mortality rates and standardised mortality ratios (SMR) were estimated. Furthermore, we examined the number and the proportion (with 95% CIs) of the 10 most common causes of death among patients with MG in each country. Only primary causes of death were analysed. The diseases as cause of death were analysed separately at category level (at the level of three characters in the ICD-10 medical coding system).
A Cox proportional hazards model was used to estimate the HR and 95% CI of death among patients with MG in relation to age, sex, country, calendar year, disease duration and comorbidity. Calendar year and disease duration were evaluated as continuous variables, while comorbidity was included as a time-varying covariate. The listed variables were prespecified. Both univariable models and multivariable models were used in the analyses. In univariable models, each preselected variable was individually present in the model. In the multivariable model, all the prespecified variables were included.
A sensitivity analysis was conducted examining the survival among patients with MG when using the PSC in comparison to the FPC.
To comply with the General Data Protection Regulation legislation, a minimum of five persons was required for separate reporting in the descriptive analyses. The final assessment of sufficient data volume was based on the estimation of the parameters, for example, the width of the CI or convergence of the estimation algorithm.
All analyses were performed using R (V.4.2.2).
Patient characteristics
Altogether, 11 136 patients with MG were identified during the study period (FPC; Denmark n=2984, Finland n=2653, Sweden n=5499). When restricting this to ≥2 diagnoses of MG during the study period, the study population was reduced to 9054 patients (PSC; Denmark n=2248, Finland n=2306, Sweden n=4500) and when further restricting to ≥2 new diagnoses of MG during the study period, the study population was reduced to 6415 patients (ISC; Denmark n=1559, Finland n=1797, Sweden n=3059) ( online supplemental figure 1 ).
Supplemental material
In the ISC, a slightly higher proportion of patients were male (51% in Denmark and Finland and 52% in Sweden), the median age at index date was 64.9 years (Q1, Q3: 49.0, 75.0) in Denmark, 64.9 years (Q1, Q3: 51.2, 74.1) in Finland and 67.5 years (Q1, Q3: 51.5, 76.8) in Sweden ( table 1 ). Most patients were diagnosed at an advanced age (≥65 years) (50% were very late onset MG in Denmark and Finland, and 55% in Sweden). Men had a higher median age at diagnosis (67.4 years; Q1, Q3: 57.9, 75.5) than women (62.6 years; Q1, Q3: 41.2, 75.6). The median follow-up time was 5.9 years (Q1, Q3: 2.4, 10.6) in Denmark, 7.3 years (Q1, Q3: 3.7, 12.6) in Finland and 7.0 years (Q1, Q3: 3.5, 11.8) in Sweden ( table 1 ).
- View inline
Baseline characteristics of patients with myasthenia gravis (MG) in Denmark, Finland and Sweden in 2000–2020 (incident subcohort)
Incidence (ISC)
The overall incidence of MG was 1.34 (95% CI 1.27 to 1.41), 1.68 (95% CI 1.60 to 1.75), and 1.62 (95% CI 1.56 to 1.68) per 100 000 during the study period in Denmark, Finland, and Sweden, respectively. The incidence of MG (per 100 000 persons) increased over time in all three countries: in Denmark from 0.86 (95% CI 0.64 to 1.14) in 2000 to 1.92 (95% CI 1.59 to 2.30) in 2020, in Finland from 1.58 (95% CI 1.26 to 1.95) in 2000 to 2.15 (95% CI 1.79 to 2.56) in 2019 and in Sweden from 0.95 (95% CI 0.76 to 1.16) in 2000 to 1.74 (95% CI 1.50 to 2.01) in 2019 ( figure 1A ).
- Download figure
- Open in new tab
- Download powerpoint
Incidence (A) and prevalence (B) of myasthenia gravis per 100 000 persons in the incident subcohort in Denmark (DK), Finland (FI) and Sweden (SE) in years 2000–2020.
The overall incidence was higher in men than in women in all three countries. In Denmark, it was 1.38 in men (95% CI 1.28 to 1.48) and 1.30 in women (95% CI 1.21 to 1.39). In Finland, the incidence was 1.74 in men (95% CI 1.63 to 1.86) and 1.61 in women (95% CI 1.51 to 1.72). In Sweden, the incidence was 1.70 in men (95% CI 1.62 to 1.78) and 1.54 in women (95% CI 1.46 to 1.62). The age-specific incidence of MG differed in men and women: in women, a higher incidence was seen in the younger age groups (patients <50 years) with a moderate increase with age, whereas a steeper increase in the incidence of MG was observed in men from 50 years onwards ( online supplemental figure 2 ).
Prevalence (PSC)
The overall prevalence of MG during the study period was 18.56 (95% CI 18.31 to 18.81) per 100 000 in Denmark, 20.89 (95% CI 20.62 to 21.16) in Finland, and 23.42 (95% CI 23.21 to 23.64) in Sweden. The prevalence of MG (per 100 000 persons) increased steadily over time in all three countries: in Denmark from 13.32 (95% CI 12.36 to 14.32) in 2000 to 25.14 (95% CI 23.88 to 26.45) in 2020, in Finland from 11.08 (95% CI 10.20 to 12.01) in 2000 to 28.52 (95% CI 27.13 to 29.95) in 2020, and in Sweden from 16.58 (95% CI 15.75 to 17.44) in 2000 to 27.18 (95% CI 26.19 to 28.19) in 2020 ( figure 1B ).
The overall prevalence of MG was higher in women than men in all three countries. In Denmark, it was 21.40 in women (95% CI 21.03 to 21.78) and 15.67 in men (95% CI 15.35 to 16.00). In Finland, the prevalence was 23.80 in women (95% CI 23.40 to 24.20) and 17.87 in men (95% CI 17.52 to 18.23). In Sweden, the prevalence was 27.27 in women (95% CI 26.94 to 27.59) and 19.56 in men (95% CI 19.28 to 19.83). The age-specific prevalence of MG increased gradually with age in both men and women ( online supplemental figure 3 ). In patient groups between 20 and 60 years of age, the prevalence was higher in women than men. In patients aged >60 years, a higher prevalence was observed in men.
Mortality (PSC)
The overall survival among patients with MG was 0.84 (95% CI 0.84 to 0.85) at 5-year follow-up, 0.70 (95% CI 0.68 to 0.71) at 10-year follow-up, 0.58 (95% CI 0.56 to 0.59) at 15-year follow-up and 0.49 (95% CI 0.48 to 0.51) at end-of-follow-up. The survival was similar in the FPC and the PSC (data not shown).
The overall SMR was 1.32 (95% CI 1.23 to 1.42) among patients with MG in Denmark, 1.23 (95% CI 1.15 to 1.33) in Finland and 1.20 (95% CI 1.14 to 1.26) in Sweden, compared with the background population. The SMR was rather stable over time in all three countries: in Denmark 1.25 (95% CI 0.82 to 1.82) in 2000 and 1.32 (95% CI 0.99 to 1.71) in 2020, in Finland 1.40 (95% CI 0.83 to 2.17) in 2000 and 1.20 (95% CI 0.90 to 1.55) in 2020 and in Sweden 1.31 (95% CI 0.99 to 1.70) in 2000 and 1.28 (95% CI 1.06 to 1.52) in 2020 ( figure 2 ).
Standardised mortality ratio of myasthenia gravis in the prevalent subcohort in Denmark (DK), Finland (FI) and Sweden (SE) in years 2000–2020.
The overall SMR was higher in women than in men in all three countries. In Denmark, it was 1.56 in women (95% CI 1.41 to 1.71) and 1.14 in men (95% CI 1.03 to 1.26). In Finland, SMR was 1.41 in women (95% CI 1.26 to 1.56) and 1.11 in men (95% CI 1.01 to 1.23). In Sweden, SMR was 1.39 in women (95% CI 1.30 to 1.49) and 1.06 in men (95% CI 0.99 to 1.13). In patients over 60 years of age, the SMR was relatively stable in men. In women over 60 years, the SMR decreased with increasing age ( online supplemental figure 4 ).
Cause of death (PSC)
In Denmark, the three most common causes of death were MG (18.10%; 95% CI 15.44% to 21.09%), chronic obstructive pulmonary disease (5.23%; 95% CI 3.79% to 7.14%) and malignant neoplasm of bronchus and lung (4.16%; 95% CI 2.89% to 5.92%) ( table 2 ). In Finland, the three most common causes of death were MG (16.87%; 95% CI 14.26% to 19.85%), chronic ischaemic heart disease (13.14%; 95% CI 10.81% to 15.87%) and acute myocardial infarction (8.58%; 95% CI 6.69% to 10.92%). Similarly, in Sweden, the three most common causes of death among patients with MG were MG (13.46%; 95% CI 11.88% to 15.22%), acute myocardial infarction (6.55%; 95% CI 5.43% to 7.87%) and chronic ischaemic heart disease (5.89%; 95% CI 4.83% to 7.16%). When combined, the most common causes of death among patients with MG in the three countries were MG (15.35%; 95% CI 14.12% to 16.67%), chronic ischaemic heart disease (6.80%; 95% CI 5.95% to 7.75%) and acute myocardial infarction (6.35%; 95% CI 5.54% to 7.28%).
Ten most common causes of death (ICD-10) among patients with myasthenia gravis in Denmark, Finland and Sweden in 2000–2020 (prevalent subcohort)
Factors associated with mortality (PSC)
When examining explanatory variables associated with the HR of death among patients with MG, the HR was found to increase with increasing age, to be similar in men and women, and to be lower in Finland (HR=0.70; 95% CI 0.63 to 0.78) and Sweden (HR=0.92; 95% CI 0.84 to 1.00) compared with Denmark ( table 3 ). Moreover, the HR of death decreased slightly over time (HR=0.97; 95% CI 0.96 to 0.98), whereas the HR of death increased with increasing duration of disease (HR=1.07; 95% CI 1.07 to 1.08). Furthermore, patients with diseases of the blood and blood-forming organs (HR=1.65; 95% CI 1.49 to 1.82), metabolic diseases (HR=1.65; 95% CI 1.51 to 1.79), thymoma (HR=1.65; 95% CI 1.65 to 2.12), respiratory diseases (HR=1.59; 95% CI 1.40 to 1.80), cancer (except thymoma) (HR=1.33; 95% CI 1.19 to 1.48), mental and behavioural disorders (HR=1.28; 95% CI 1.14 to 1.45) and diseases of the circulatory system (HR=1.25; 95% CI 1.15 to 1.36) had an increased HR of death compared with patients without these comorbidities. In contrast, there was no increased risk of death among patients with MG with other autoimmune diseases, ophthalmological diseases, musculoskeletal diseases or severe infections.
Cox proportional hazards model examining the HR of death and 95% CIs among patients with myasthenia gravis in Denmark, Finland and Sweden in relation to explanatory variables in 2000–2020 (prevalent subcohort)
In this large population-based cohort study, we identified more than 9000 patients with ≥2 diagnoses of MG in Denmark, Finland and Sweden over a 20-year study period. The incidence of MG increased over time in all three countries. In younger age groups, a higher incidence was observed in women than men, which increased moderately with age. While in men, a steeper increase with age was observed from age 50 years onwards. The prevalence of MG increased steadily over time in all three countries. A higher prevalence was found in women than in men in the younger age groups. In women, the prevalence increased moderately with age, while in men a steep increase was seen from age 50 years. The SMR was increased in patients with MG in all three countries, especially in women.
Our study showed an overall incidence of MG of 1.34 in Denmark, 1.68 in Finland and 1.62 per 100 000 in Sweden. These estimates are higher than earlier reports from Norway and Denmark (0.4 per 100 000), but slightly lower than those previously reported in Sweden (2.9 per 100 000) and more recently in Germany (4.6 per 100 000). 9–11 22 Worldwide, the mean incidence of MG has been reported as 1.0 (range: 0.3–2.8) per 100 000. 8 We found that the incidence of MG increased over time, similarly to a Swedish study 10 but in contrast to a Danish study. 9 The overall prevalence of MG in this study was 18.56 in Denmark, 20.89 in Finland and 23.42 per 100 000 in Sweden. These estimates align with previous studies, in particular from Finland and Sweden. 12 13 However, the estimates diverge from the findings of an earlier Swedish study (36.1/100 000), 10 a German study (39.3/100 000) 11 and a literature review (10/100,000). 8 In line with our findings, an earlier study from Denmark found an increasing prevalence of MG over time. 9 The observed discrepancies in incidence and prevalence could result from differences in study periods, data sources or definitions of MG, but in general, the increasing incidence and prevalence could be the result of, for example, the rise in the incidence of autoimmune diseases, ageing of the population, increased life expectancy, improved diagnostics and access to testing, improvement of the quality of register data over time, and to a more minor extent, reduced mortality over time.
The observed mortality of MG is largely in line with two Danish studies, which reported overall survival of 81% and 69% at 5 and 10 years of follow-up, respectively, 23 and an overall SMR of 1.41. 16 Similarly, a Swedish study reported a mortality ratio of 1.28 in patients with MG compared with the general population. 17 The most common causes of death among patients with MG in the three countries were MG, chronic ischaemic heart disease and acute myocardial infarction, supporting the findings of previous studies 17 23 and aligning also with the general population in the three studied countries. 24 Additionally, this study analysed information about the underlying cause of death rather than the immediate cause. Moreover, due to limitations in data capture within the registry, it is not possible to further substantiate the underlying pathology attributing MG as the cause of death. This likely encompasses MG-related causes such as respiratory failure, pneumonia or treatment-related comorbidities like diabetes and cardiovascular disease. The overall SMR was markedly higher in women than men in all three countries, as reported previously in a Danish study. 16 This could reflect the earlier age at diagnosis in women or possibly suboptimal management of MG in women. Further reasons for the higher risk of death in women could include the more severe and early onset forms of the disease, which are more common in women and women being more prone to autoimmune diseases in general. In addition, the early-onset form of MG results in a longer period of treatment over lifetime and, thus, accumulation of the long-term consequences of the treatments. Comorbidity patterns specific to early-onset MG could play a role too, especially other autoimmune diseases. 25 As opposed to the SMR which is a relative measure, the absolute overall HR of death was however similar in men and women.
When examining factors associated with the risk of death among patients with MG, we observed that the risk of death increased with increasing age and duration of disease but decreased slightly over calendar time. Moreover, patients with MG with diseases of the blood and blood-forming organs, respiratory diseases, metabolic diseases and thymoma had an increased risk of death. Patients with cancer (except thymoma), diseases of the circulatory system and mental disorders also had a moderately increased risk of death. Nevertheless, and in contrast to the general population, 26–28 there was no difference in the risk of death among men and women with MG, which may reflect the adjustment for important predictors of death among patients with MG. Severe infections can be common in undertreated MG, but in this study, they were not associated with the risk of death, potentially indicating the level of public healthcare with universal coverage in the three studied countries.
The main strength of this study is the real-world setting with complete nationwide coverage in three Nordic countries with minimal loss to follow-up. As this was a register study, it was not possible to compare the results to, for example, data from clinical centres treating patients with MG, but the Nordic registries have been shown to be of great validity regarding data coverage and accuracy. 29–31 All citizens are included in the health and population registries regardless of social status, income or insurance. Therefore, the data are representative of the entire population with a negligible selection bias, offering a major benefit compared with cohorts that rely on clinical, insurance or survey data. Furthermore, all Nordic countries have publicly funded, universal and high-quality healthcare available to all citizens. Accessing data from more than one country allows the evaluation of broader trends and cross-country differences. However, study limitations include the country-specific differences in the registry structures and data recording practices. For transparency, we report results separately for the three countries. Additionally, case identification was based on ICD codes from specialised inpatient and outpatient care, thus not including primary healthcare. Given that MG is not typically diagnosed or managed by general practitioners, we deem the risk of missing true cases small. To increase the specificity of MG diagnosis, we required ≥2 separate diagnoses from the registries. Due to data constraints, specifically the absence of primary healthcare data and confirmatory MG diagnoses, we are unable to ascertain the age at onset and diagnostic delay. Nevertheless, our findings align with the established age distribution at diagnosis between men and women. Finally, the observational nature of the study design implies that causality cannot be readily inferred from this study.
In conclusion, this population-based study from three Nordic countries showed that the incidence and prevalence of MG increased from 2000 to 2020, whereas the mortality was stable over time in Denmark, Finland and Sweden. The findings of this study highlight the importance of careful management of patients with MG and the need for further improved care, especially in the younger age groups of women.
Ethics statements
Patient consent for publication.
Not applicable.
Ethics approval
This study was approved by Statistics Denmark (708396), the Finnish Data Permit Authority, Findata (THL/1010/14.02.00/2021), Statistics Finland (TK/2457/07.03.00/2021), and the Swedish National Board of Health & Welfare (Socialstyrelsen; Dnr 16836/2021). Ethical approval was obtained by the Swedish Ethics Review Authority (Dnr 2021-00858). Ethical approval was not required according to Finnish and Danish legislation, given that the study was observational and based solely on pseudonymised registry data.
Acknowledgments
The authors would like to thank Karoline Doser, PhD, Mirkka Koivusalo, PhD, and Simone Møller Hede, MSc for project management and other support in the study conduct, Harlan Barker, MSc, for language review (MedEngine Oy and MedEngine DK ApS), and Didier Pitsi, PhD (UCB Pharma SA) for reviewing this manuscript. Preliminary findings of this study were presented at the 9th Congress of the European Academy of Neurology in Budapest, Hungary 1 to 4 July 2023.
- Gilhus NE ,
- Tzartos S ,
- Evoli A , et al
- Phillips WD ,
- Meriggioli MN ,
- Romi F , et al
- Verschuuren JJ
- Guptill JT ,
- Meriggioli MN
- Deenen JCW ,
- Horlings CGC ,
- Verschuuren JJGM , et al
- Somnier FE ,
- Keiding N ,
- Westerberg E ,
- Biskup J , et al
- Sveinsson O ,
- Thormar G , et al
- Sipilä JOT ,
- Soilu-Hänninen M ,
- Rautava P , et al
- Dresser L ,
- Wlodarski R ,
- Rezania K , et al
- Phillips LH ,
- Hansen JS ,
- Danielsen DH ,
- Somnier FE , et al
- ↵ Befolkningstal . 2023 . Available : https://www.dst.dk/da/Statistik/emner/borgere/befolkning/befolkningstal
- Statistics Finland
- Statistiska Centralbyrån
- Narayanaswami P ,
- Sanders DB ,
- Wolfe G , et al
- Christensen PB ,
- Jensen TS ,
- Tsiropoulos I , et al
- World Health Organization
- Laakso SM ,
- Myllynen C ,
- Strbian D , et al
- Statistics Denmark
- Tilastokeskus
- Socialstyrelsen
- Smith Jervelund S ,
- De Montgomery CJ
- Maret-Ouda J ,
- Wahlin K , et al
Supplementary materials
Supplementary data.
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
Contributors Substantial contributions to the conception and design of the work: JV, SA, MS, JM, TY-o, IL-S, FB and FP. Substantial contributions to the analysis of the data: JM. Substantial contributions to the interpretation of the data: all authors. Drafting, reviewing and revising the manuscript critically for important intellectual content: all authors. Final approval of the version to be published: all authors. Agree to be accountable for all aspects of the work in ensuring that questions related to accuracy or integrity of any part of the work are appropriately investigated and resolved: all authors. TY is the guarantor.
Funding The study was funded by UCB Pharma.
Competing interests JV: Roche, Sanofi Genzyme, Sarepta Therapeutics, Novartis Pharma AG, Fulcrum Therapeutics, Biogen, Lupin, Amicus, Regeneron, Argenx BVBA, UCB Biopharma SPRL, Arvinas, ML Biopharma, Atamyo Therapeutics, Horizon Therapeutics, Dyne Therapeutics, Alexion Pharmaceuticals, Edgewise Therapeutics, Genethon, Reneo Pharma, Pharnext, Janssen Pharmaceutical, Khondrion, Dynacure SAS. SA: Merck, Roche, Biogen, Novartis, UCB Pharma, Lundbeck. MS was previously an employee of UCB Pharma, Espoo, Finland. JM is an employee of MedEngine Oy, Finland. LM was previously an employee of MedEngine DK ApS, Denmark. TBO is an employee of MedEngine DK ApS, Denmark. TY-o is the owner of MedEngine Oy and MedEngine DK ApS. IL-S is an employee of UCB Pharma, Stockholm, Sweden. FB is an employee and stockholder of UCB Pharma, Copenhagen, Denmark. FP: Janssen, Merck KGaA, UCB, Chugai, Lundbeck, Roche, Novartis.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Read the full text or download the PDF:
COMMENTS
Myasthenia gravis (MG) is the most common disorder affecting the neuromuscular junction (NMJ) of the skeletal muscles. The classic presentation is a fluctuating weakness that is more prominent in the afternoon. It usually involves muscles of the eyes, throat, and extremities. The reduced transmission of electrical impulses across the neuromuscular junction due to the formation of ...
Myasthenia gravis (MG) is an autoimmune neurological disorder characterized by defective transmission at the neuromuscular junction. The incidence of the disease is 4.1 to 30 cases per million person-years, and the prevalence rate ranges from 150 to 200 cases per million. MG is considered a classic example of antibody-mediated autoimmune ...
Myasthenia gravis is an autoimmune disease in which antibodies bind to acetylcholine receptors or to functionally related molecules in the postsynaptic membrane at the neuromuscular junction. The ...
Myasthenia gravis (MG) is an autoimmune disease caused by antibodies against the acetylcholine receptor (AChR), muscle-specific kinase (MuSK) or other AChR-related proteins in the postsynaptic ...
A panel of 15 international experts in the treatment of MG was convened and, in 2016, published an international consensus guidance for the management of MG. 1. Results of several new trials of MG treatment have been published since that guidance statement was published, and in 2019, the panel reviewed the previous recommendations for currency ...
Myasthenia gravis (MG) is an autoimmune neurological disorder characterized by defective transmission at the neuromuscular junction. The incidence of the disease is 4.1 to 30 cases per million person-years, and the prevalence rate ranges from 150 to 200 cases per million. MG is considered a classic example of antibody-mediated autoimmune disease. Most patients with MG have autoantibodies ...
1. Introduction. Myasthenia Gravis (MG) is an autoimmune disorder mediated by autoantibodies and causing fluctuating skeletal muscle weakness. It is a disease of the neuromuscular junction, in which autoantibodies to the acetylcholine receptor (AChR), muscle specific kinase (MuSK) or rarely other antibodies directed against the neuromuscular junction proteins cause fatigable muscle weakness [1 ...
Myasthenia gravis (MG) is an autoimmune disorder that affects the neuromuscular junction, leading to muscle weakness and fatigue. ... Article Google Scholar Melzer, N. et al. Clinical features ...
Myasthenia gravis (MG) is the most common autoimmune disorder that affects the neuromuscular junction. MG is largely a treatable disease but can result in significant morbidity and even mortality. This can usually be prevented with a timely diagnosis and appropriate treatment of the disease.
Myasthenia gravis (MG) is a rare, autoimmune, antibody-mediated, neuromuscular disease. ... Article Google Scholar Myasthenia Gravis Foundation of America. Intravenous immunoglobulin (IVIg) (2018).
Disorders of neuromuscular transmission can be of immunological, toxic or genetic origin. Among these rare disorders, myasthenia gravis is the most frequent. The clinical hallmark of myasthenia (MG) gravis is a fluctuating pronounced weakness limited to the voluntary muscles. Characteristically, muscular exertion increases the myasthenic weakness.
The diagnosis of autoimmune Myasthenia Gravis (MG) remains clinical and rests on the history and physical findings of fatigable, fluctuating muscle weakness in a specific distribution. Ancillary bedside tests and laboratory methods help confirm the synaptic disorder, define its type and severity, classify MG according to the causative antibodies, and assess the effect of treatment objectively.
Myasthenia gravis (MG) is a rare, autoimmune neuromuscular junction disorder. Contemporary prevalence rates approach 1/5,000. MG presents with painless, fluctuating, fatigable weakness involving specific muscle groups. Ocular weakness with asymmetric ptosis and binocular diplopia is the most typical initial presentation, while early or isolated oropharyngeal or limb weakness is less common ...
Background Myasthenia gravis is a neuromuscular autoimmune disorder characterized by weakness and disability in the voluntary muscles. There have been several preliminary studies on the epidemiology of myasthenia gravis in different parts of the world and the effectiveness of common drugs in its treatment, but there has been no comprehensive study of the efficacy of common drugs in the ...
The approval of rozanolixizumab represents an advancement in therapy for generalized myasthenia gravis. The provision of individualized, targeted, and well-tolerated treatment is valuable for the patients whose myasthenia gravis is not well controlled and who are seeking a medication with a rapid onset of action to improve their symptoms and overall quality of life.
Myasthenia gravis (MG) is a debilitating autoimmune neuromuscular disease characterised by fluctuating muscle weakness with a prevalence of 100-350 people per million globally [1, 2].MG causes neuromuscular or muscle fatiguability, where patients' symptoms worsen with sustained activity [].However, fatigue in MG goes beyond neuromuscular fatiguability and is also characterised by ...
Myasthenia gravis is an autoimmune neuromuscular disease that is driven by pathogenic autoantibodies against the nicotinic acetylcholine receptor or muscle-specific tyrosine kinase (MUSK).1 Long-term treatment is essential for most patients and must be tailored to the particular type of myasthenia gravis. In the past 5 years there has been a surge in the development of targeted molecular ...
Immune-mediated myasthenia gravis (MG) is a rare and serious neurologic adverse event that has been associated with ICIs requiring prompt treatment. In the Jehovah's Witness population, typical management of these adverse events may not be options, and alternative treatment choices would be needed.
Objective: The aims of this article are to review the clinical aspects of rozanolixizumab, to describe clinical trial results that led to the drug's approval, and to examine the impact on patient care to aid clinical decision making. Data sources: A PubMed search was conducted using the terms Rystiggo™, rozanolixizumab, rozanolixizumab therapy, and myasthenia gravis.
Myasthenia gravis patients displayed distinct clinical symptoms, with ptosis being the most common (87.9%). Myasthenia Gravis Foundation of America classification indicated the highest proportion in subgroup IIa (24.2%), with myasthenia gravis predominating in limb and axial muscles (Group a) observed in 51.5% of cases. Needle electromyography ...
Myasthenia gravis (MG) is an autoimmune disease mediated by antibodies and with the participation of complement, in which antibodies bind to acetylcholine receptors or functionally related ...
1 Introduction. Myasthenia gravis (MG) is an autoimmune disorder affecting the neuromuscular junction, primarily instigated by the presence of antibodies targeting various postsynaptic components ().Among these antibodies, anti-acetylcholine receptor antibodies (AChR-Ab) stand as the most prevalent, while antibodies against MuSK (MuSK-Ab), Lrp4, and agrin are comparatively less common ().
Introduction. Myasthenia gravis (MG) is a rare acquired autoimmune disorder of the neuromuscular junction (NMJ), caused by antibodies that target the post-synaptic membrane ().These antibodies commonly are to the nicotinic acetylcholine receptor (AChR) but in a smaller proportion of cases, antibodies to muscle specific tyrosine kinase (MuSK) or to lipoprotein receptor-related protein 4 (Lrp-4 ...
Recommendations for the treatment of myasthenia gravis (MG) have been difficult to develop because of limited evidence from large randomized controlled trials. New drugs and treatment approaches have recently been shown to be effective in phase 3 studies in seropositive generalized (g) MG. One such drug is efgartigimod, a human-Fc-fragment of IgG1, with a high affinity for the endosomal FcRn ...
Table 1 shows the item scores for the Myasthenia Gravis-Activities of Daily Living (MG-ADL) score, whereas Table S1 presents the arterial blood gas analysis at various time points. He was hospitalized and received two cycles of chemotherapy prior to thymectomy. Histopathological examination showed invasive thymoma, type B2 and the disease stage ...
@article{Zhou2024ScreeningFI, title={Screening for immune-related biomarkers associated with myasthenia gravis and dilated cardiomyopathy based on bioinformatics analysis and machine learning}, author={Guiting Zhou and Shushu Wang and Liwen Lin and Kachun Lu and Zhichao Lin and Ziyan Zhang and Yuling Zhang and Danling Cheng and KaMan Szeto and ...
Background Incidence and prevalence rates of myasthenia gravis (MG) vary considerably across studies, and mortality risk is rarely addressed. We examined the prevalence and incidence rates, mortality and factors associated with mortality with MG. Method This was a registry linkage study based on nationwide health and administrative registries of Denmark, Finland and Sweden (populations of 5.9 ...